
Abstract
Summary: This report documents a case of angiotropic large cell lymphoma (ALCL) with imaging characteristics of CNS vasculitis. A 47-year-old man was unresponsive after a 5-month progression of neurologic deterioration and intermittent fevers. MR imaging revealed multiple areas of abnormally increased T2 signal intensity in the cerebral cortex and subcortical white matter. Despite corticosteroid treatment for presumed CNS vasculitis, the patient died. Necropsy revealed a diffuse intravascular malignant mononuclear proliferation consistent with ALCL.
CNS vasculitis is characterized by inflammatory damage to blood vessel walls, associated vascular thrombosis, and ischemic damage to the tissue served by the affected vessels. The progressive clinical neurologic abnormalities in CNS vasculitis are often associated with the neuroradiologic findings of multiple brain parenchymal lesions consistent with ischemic damage on CT scans and MR images. Angiotropic large cell lymphoma (ALCL), an aggressive intravascular neoplasm, can mimic these imaging findings.
Case Report
A 47-year-old man with a 3-month history of intermittent numbness and tingling in his right arm, episodes of transient aphasia, headaches, memory loss, and decreased motor coordination was treated with warfarin in early September 1999 for presumed cerebral ischemia. Three weeks later, he reported fatigue and lethargy and was found to be severely anemic (hemoglobin, 7.8 g/dL). Anticoagulation was discontinued, and findings from an endoscopic examination for an occult source of gastrointestinal bleeding were negative. Results for a workup for anemia, including blood cultures, HIV testing, and a Coomb’s antiglobulin test were negative. Serum levels of lactate dehydrogenase, thyroid stimulating hormone, cortisol, folate, vitamin B12, lead, haptoglobulin, and C3 and C4 complement levels were within normal limits. Tests for antinuclear antibodies and antineutrophil cytoplasmic autoantibodies (p-ANCA and c-ANCA) were negative as well. One week later, on October 8, the patient was admitted to an outside institution with a temperature of 104°F, shaking chills, and tachypnea. A ventilation-perfusion scan revealed no evidence for a pulmonary embolus, and a chest CT scan showed no evidence of a lung mass. The patient’s erythrocyte sedimentation rate (ESR) was elevated, ranging from 60–90 mm/h. No cause for the patient’s symptoms was identified, and he was discharged after stabilization.
The patient underwent additional outpatient diagnostic testing, including cerebral angiography on October 22, which was negative for vasculitis in either the carotid or vertebrobasilar arterial systems. Head MR imaging performed 1 day later revealed focal areas of increased T2 signal intensity in the central pons and at the junction between the anterior limb of the left internal capsule and the globus pallidus. Two weeks later, the patient was readmitted with increased confusion, headaches, lower extremity paresis, and gait instability. A non-contrast-enhanced head CT scan performed at the time of his admission on November 7 showed no evidence of acute intracranial hemorrhage or mass effect. Despite empiric treatment with corticosteroids, his neurologic status deteriorated. By the fourth day of his hospitalization, the patient became unresponsive and was transferred to our institution on November 10.
At transfer, the patient had mild hypotension (90 mmHg/50 mmHg) and a low-grade fever (100.4°F). At neurologic examination, the patient withdrew extremities and opened his eyes to painful stimuli but demonstrated no purposeful movements. His pupils were reactive but sluggish. The patient’s physical examination was otherwise normal. No adenopathy was detected. He was found to be severely anemic (hemoglobin, 6.8 g/dL) and mildly thrombocytopenic (platelet count, of 139,000/mm3). Blood cultures were negative, but his ESR remained elevated at 94 mm/h. CSF was normal.
A head CT scan acquired the day of his admission showed a hypoattenuated wedge-shaped area encompassing both the cortex and subcortical white matter of the left frontoparietal junction without associated hemorrhage or mass effect (Fig 1). MR imaging, performed with and without contrast agent administration during the third day of hospitalization, showed a corresponding hyperintense T2 lesion in this region and additional lesions also involving cortical, subcortical, and deep gray and white matter structures (temporal lobes bilaterally, central pons, right middle cerebellar peduncle, and corpus callosum) (Fig 2A, C, and E). Diffusion-weighted images (Fig 2B and D) showed correlative signal intensity changes in most areas of T2 abnormality consistent with a component of either acute or subacute ischemic change. Several areas of abnormal diffusion showed no correlative T2 signal intensity abnormalities (Fig 2C and D) consistent with areas of acute ischemic change. Except for the bilateral pontine lesions, each of the supratentorial lesions and also those lesions involving the right middle cerebellar peduncle appeared to have developed in the interval since his previous MR study obtained 21 days earlier. T1-weighted contrast-enhanced images depicted subtle enhancement of the left frontoparietal region, with a slight degree of sulcal effacement in the same area. No enhancement or mass effect was observed in association with the other lesions. Repeat non-contrast-enhanced axial T2-weighted fluid-attenuated inversion recovery MR images performed on November 17 revealed additional signal intensity abnormalities in the right frontal region as well as the progression of lesions adjoining the atria of the lateral ventricles (Fig 3) consistent with additional areas of ischemic damage.

Images obtained on November 10, 1999.
Non-contrast-enhanced head CT depicts a wedge-shaped low-attenuation area (arrowhead) involving both the gray and white matter of the superficial anterior left parietal region most consistent with an ischemic focus.
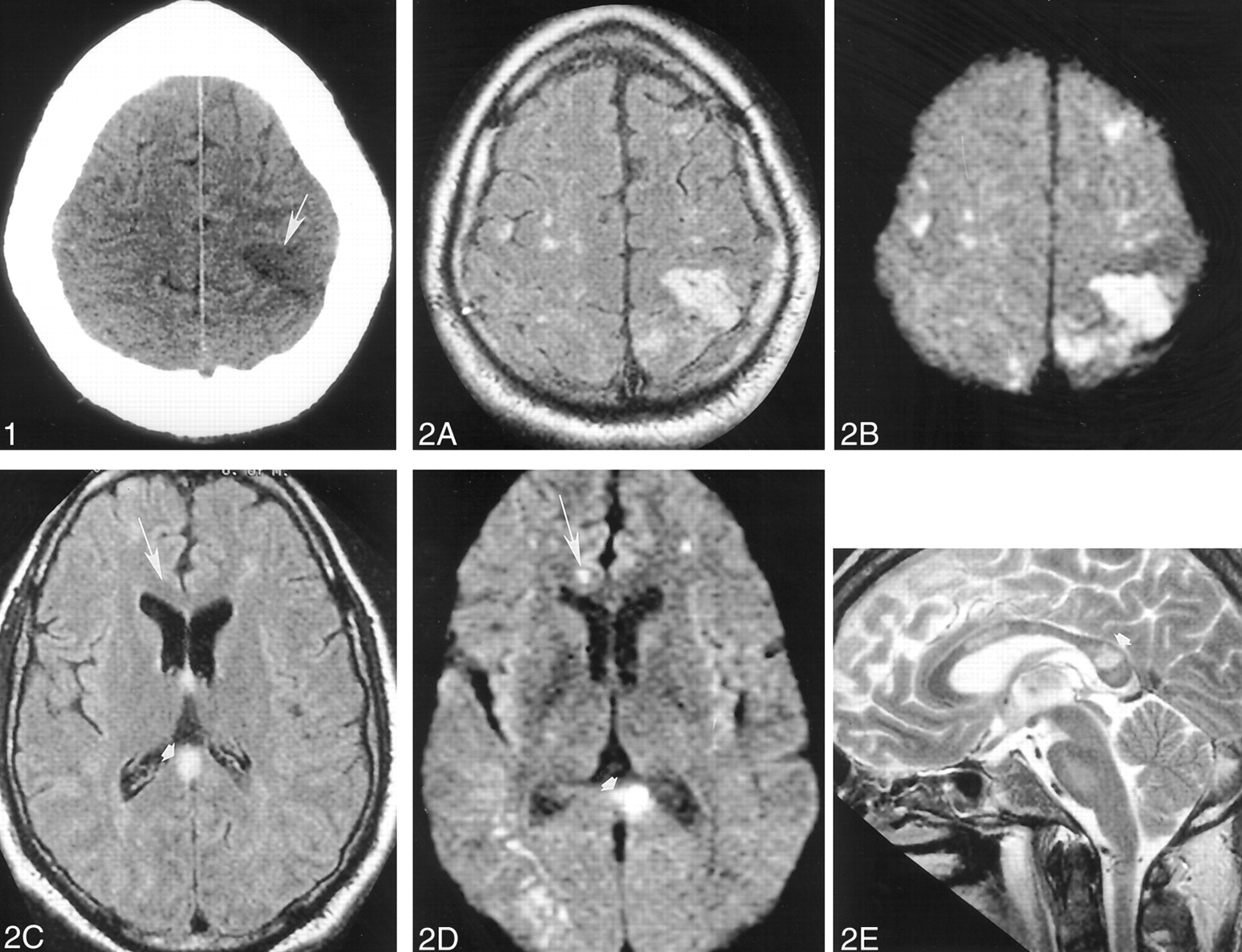
Images obtained on November 12, 1999.
A–E, Non-contrast-enhanced axial fluid-attenuated inversion recovery (10,002/105 [TR/TE]) (A and C), axial echo-planar diffusion-weighted (10,000/105) (B and D), and sagittal T2-weighted (3000/90) (E) head MR images depict multiple cortical and subcortical lesions. Most of these lesions are better seen on diffusion-weighted images and are therefore most consistent with areas of acute or subacute ischemic change (arrow). Note the lesion (arrowhead) in the splenium of the corpus callosum.

Images obtained on November 17, 1999.
A and B, Repeat non-contrast-enhanced axial fluid-attenuated inversion recovery (10,002/105) head MR images disclose interval development of new areas of abnormal signal intensity in the right frontal region (arrowhead) and the genu of the corpus callosum (arrow) over 5 days. These findings are consistent with additional areas of ischemic change.
On the basis of clinical and imaging data, a tentative diagnosis of CNS vasculitis was made. However, despite treatment with high-dose corticosteroids (240 mg prednisone/day), the patient’s condition continued to worsen. He was scheduled for brain biopsy but died 13 days after admission from complications of nosocomial pneumonia.
Autopsy revealed diffuse intravascular B cell infiltration consistent with ALCL. Histologic examination of the brain revealed areas of focal edema with extensive occlusion of cerebral vessels by B cell lymphoma (Fig 4A). Multiple bilateral petechial hemorrhages and small white and gray matter areas of frank infarction were also identified (Fig 4B). The pituitary gland vasculature also contained neoplastic lymphocytes as well. Neoplastic B cells were also identified in the blood vessels of the lungs, spleen, liver, kidneys, thyroid, pancreas, and prostate. A bone marrow specimen was negative for lymphoma.
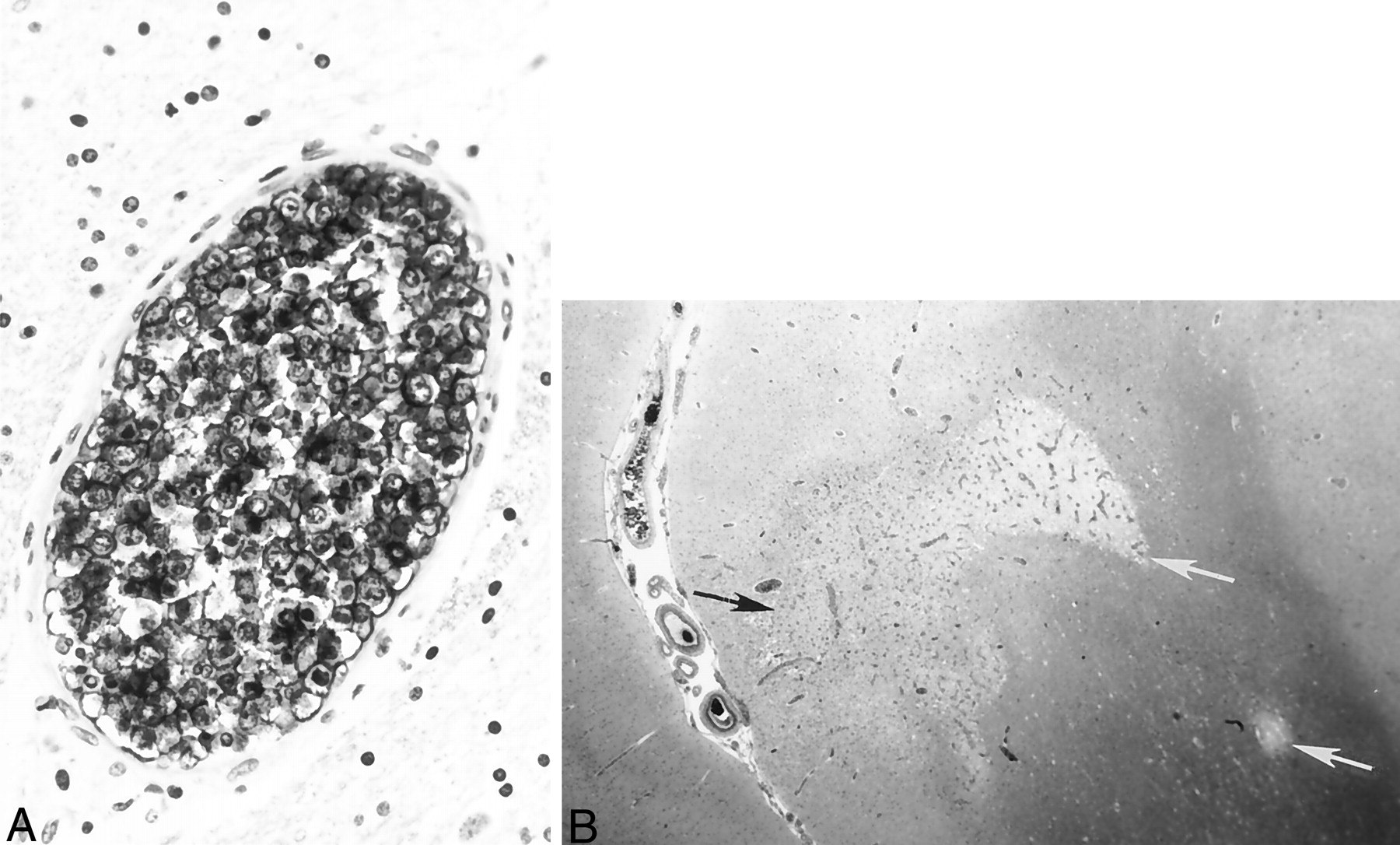
Histologic specimens confirming ALCL.
A, Immunostaining with L26, a monoclonal antibody B cell marker, reveals large, atypical intravascular tumor cells occluding the lumen of a small cerebral blood vessel. Note the large ratio of vessel diameter to wall thickness, indicative of neoplastic lymphocytes occluding and distending the vessel lumen. In contrast to a true vasculitic process, the vessel wall itself does not show inflammatory damage.
B, Hematoxylin and eosin staining shows gray (black arrow) and white (white arrows) matter infarctions resulting from blood flow compromised by intravascular lymphoma.
Discussion
ALCL, also referred to as intravascular lymphomatosis or malignant angioendotheliomatosis, is a rare yet aggressive type of non-Hodgkin’s lymphoma. It is characterized by a proliferation of neoplastic cells within the lumen of small arteries, arterioles, capillaries, and venules. Vascular occlusion results in tissue infarction. Although the microangiopathy associated with ALCL may involve multiple organ systems, the blood vessels of the CNS and skin are most commonly affected. ALCL must be distinguished from a variety of vasculitic, neoplastic, and thromboembolic processes in which vascular damage results in ischemic injury to the tissue served by the affected vessels. The clinical manifestations of vasculitides that involve the CNS may mirror the multifocal and rapidly progressive neurologic abnormalities in ALCL that include headaches, mental status changes, cerebrovascular events, spinal cord or nerve root syndromes, subacute encephalopathies, cranial or peripheral neuropathies, seizures, and delirium. ALCL has been known to clinically mimic primary angiitis of the CNS (PACNS) and diffuse systemic necrotizing vasculitis such as polyarteritis nodosa (1–6). ALCL and PACNS primarily affect small parenchymal and leptomeningeal vessels and result in multifocal ischemic lesions that are visible on radiologic images. Both have a rapid course and may respond to corticosteroid therapy, although the response is almost always transient in ALCL (1). Constitutional signs and symptoms such as fever, malaise, weight loss, and arthralgias are common in ALCL and systemic necrotizing vasculitis, but are usually absent in PACNS (3).
To our knowledge, Domizio et al (7) have published the only comprehensive review of ALCL since its initial description in 1959 by Pfleger and Tappeiner. Of the 79 fully characterized cases discussed by Domizio et al, 32% of the patients had symptoms attributable to CNS lesions alone, whereas 12% of the patients had CNS and systemic symptoms (7). To date, only sporadic descriptions of the neuroimaging features of ALCL have appeared in the literature. The current report describes a case of ALCL with the imaging features and clinical characteristics of CNS vasculitis.
Although neuroimaging features in ALCL and vasculitis may be identical, the radiologic findings are nonspecific for both disease processes. Cranial CT scanning has been reported to disclose multifocal hypodense lesions in up to 86% of histologically confirmed ALCL cases (8). However, because of the numerous reports of normal CT scans acquired in patients with neurologic manifestations of ALCL (6–10), CT is generally considered to be nondiagnostic. ALCL follows a rapidly progressive worsening of clinical course, and serial CT scans may show the development of multiple new cerebral infarctions or progression of existing lesions (1, 2, 11).
Cranial MR images acquired in patients with ALCL involving the CNS may be normal (1, 4, 8) or may show nonspecific hyperintense white matter lesions (1, 5, 8, 9, 10) suggestive of small vessel ischemic disease or demyelination. In ALCL as well as in CNS vasculitis, multiple areas of increased signal intensity appear in the white and gray matter bilaterally on T1-, intermediate-, and T2-weighted images. Although such lesions may occur diffusely throughout the brain in both processes, white matter lesions in CNS vasculitis commonly occur in the deep white matter, and those in ALCL often occur in the subcortical and periventricular white matter (1, 12). Although non-contrast-enhanced MR images may be of limited use in identifying lesions previously undisclosed by CT studies, spinal and cranial contrast-enhanced T1-weighted MR images have increased the sensitivity by which brain and spinal cord lesions can be identified, monitored, and targeted for biopsy (13). Similar to lesions visualized on CT scans, variable patterns of meningeal and parenchymal contrast enhancement may exist. Contrast-enhanced MR images can clearly delineate small areas of enhancement and show such lesions as distinct from surrounding hypoattenuated areas of edema (13). Other lesions associated with ALCL, however, may not enhance or show only subtle enhancement, as was the case in our patient (1, 6, 8, 9).
Cerebral angiography reveals multivessel segmental stenoses and dilatations that appear in abnormal beadlike patterns consistent with CNS vasculitis in as many as 45% of cases of ALCL with CNS manifestations (8). Cerebral angiography has been shown to help detect vasculitic lesions not visible on MR images (12). However, numerous reports of normal angiograms in ALCL with CNS involvement exist in the literature, further underscoring the need for a definitive biopsy in patients with presumed CNS vasculitis who have normal cerebral angiographic findings (1, 2, 8, 9).
In the current case, the patient’s clinical history, neuroimaging studies, and the absence of any physical or laboratory abnormalities specific to any other known disease process supported a preliminary clinical diagnosis of CNS vasculitis. Before a brain biopsy could be performed, the patient died, and the diagnosis of systemic ALCL was made at autopsy. Because no pathognomonic clinical findings of ALCL exist, an antemortem diagnosis of the disease is difficult. Approximately 50% of ALCL cases are diagnosed at autopsy (7). Nonspecific laboratory abnormalities that often accompany ALCL include all or one of the following: anemia, increased serum lactate dehydrogenase levels, or an elevated ESR. Elevated ESR occurs in approximately 80% of patients with neurologic manifestations of ALCL (8, 9). Despite the intravascular location of the malignant cells, circulating neoplastic cells are rarely evident at examination of peripheral blood, bone marrow, or CSF. CSF analysis may show a mild leukocytosis or an increased amount of protein, but cytologic results are negative for either finding in most patients. In contrast to other hematologic malignancies, involvement of the superficial lymph nodes is usually minimal or absent.
ALCL is a multisystemic disease that can affect any organ and is understood as generalized at the time of initial diagnosis. Dissemination of the disease is rapid and extensive such that the average survival after antemortem diagnosis is approximately 5 months (7). Although the disease can have protean manifestations depending on the affected organs, neurologic and cutaneous involvement is most common. Neuroimaging studies may suggest a clinical diagnosis of cerebral vasculitis, but because no markers or noninvasive diagnostic tests for ALCL exist, a tissue biopsy of an involved organ remains the only means of a definitive antemortem diagnosis. Although ALCL is generally fatal, reports of clinical improvement and even remission after combination chemotherapy appear in the literature (14).
Conclusion
The potential to improve the clinical course of ALCL is contingent on an accurate and early diagnosis and underscores the need to consider ALCL in the differential diagnosis of CNS vasculitis and to aggressively pursue diagnostic efforts in patients with progressive multifocal systemic deficits or CNS deficits or both.
References
- Received March 28, 2001.
- Accepted after revision August 9, 2001.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Vasculitis on brain angiography is not always vasculitis: intravascular large B-cell lymphoma mimicking central nervous system vasculitis
- Recurrent cerebrovascular accidents caused by intravascular lymphoma in a dog
- Clinical Reasoning: A 66-year-old man with recurrent multi-territory infarcts
- Intravascular Lymphoma: The Oncologist's "Great Imitator".