
Abstract
Summary: We report on a case of Wegener granulomatosis in the pterygopalatine fossa that was associated with trigeminal neuropathy. MR and CT examinations were useful in depicting the extent of the lesion and suggesting a perineural spread. Diagnosis was confirmed with positive serum assay findings for the presence of cytoplasmic antineutrophil cytoplasm antibody.
Wegener granulomatosis (WG) is a rare, disseminated, granulomatous disease of unknown origin. It primarily involves the upper and lower respiratory tracts with or without glomerulonephritis and systemic vasculitis (1). Trigeminal neuropathy is rarely recognized as the sole presenting sign (1, 2). We report on a case of WG involving a pseudotumoral lesion of the pterygopalatine fossa (PPF) that was associated with trigeminal neuropathy. CT and MR imaging were useful in depicting the lesion in the PPF. When a lesion is discovered in the PPF and when histopathologic analysis yields only nonspecific inflammatory findings, the diagnosis of WG should be suspected and verified with a cytoplasmic antineutrophil cytoplasm antibody (c-ANCA) test (3, 4).
The PPF is a small space delineated by the posterior wall of the maxillary sinus and the anterior surface of the pterygoid process of the sphenoid bone. The PPF connects directly with six locations: the infratemporal fossa, via the long pterygomaxillary fissure; the nasal cavity, via the small sphenopalatine foramen; the apex of the orbit, via the inferior orbital fissure; the palate, via the greater and lesser palatine canals; the Meckel cave, via the foramen rotundum; and the petrous apex, via the pterygoid canal. From the infraorbital canal, the infraorbital nerve passes through the PPF to form the maxillary nerve, which passes through the foramen rotundum. The palatine nerve extends inferiorly from the PPF through the pterygopalatine canal to the greater and lesser palatine foramina, reaching the mucosal surface of the hard palate (5).
Case Report
A 49-year-old man with a 2-year history of chronic rhinosinusitis presented with subacute profound hypesthesia of the left palate and ipsilateral lip and paresthesias of the left cheek and left ocular proptosis. At examination, rhinosinusitis was present without other cranial nerve abnormalities. The erythrocyte sedimentation rate was 45 mm/h. The white blood cell count and serum creatinine levels were normal. No urine abnormalities were found. The chest radiograph was normal. Lumbar puncture showed no cellular reaction. A CT scan (Fig 1A) showed mucosal thickening of the paranasal sinuses and a focal bony lysis of the posterior wall of the left maxillary sinus. The vertical part of the left palatine bone appeared demineralized. The left foramen rotundum was asymmetric and seemed enlarged. MR imaging (Fig 1B–D) showed an enhancing soft-tissue mass within the PPF, encasing the maxillary artery and extending into the inferior orbital fissure. Pre- and postcontrast T1-weighted images showed a tissular proliferation obliterating the fatty tissue of the foramen rotundum. Biopsy of the PPF, performed via a transbuccal approach, revealed nonspecific inflammatory changes with infiltration of small lymphoid, neutrophilic, and histiocytic cells, suggestive of a pseudotumor. A c-ANCA assay revealed a positive titer of 1:300 (negative, below 1:80), confirming the diagnosis of WG. No other manifestation of granulomatous disease was found.
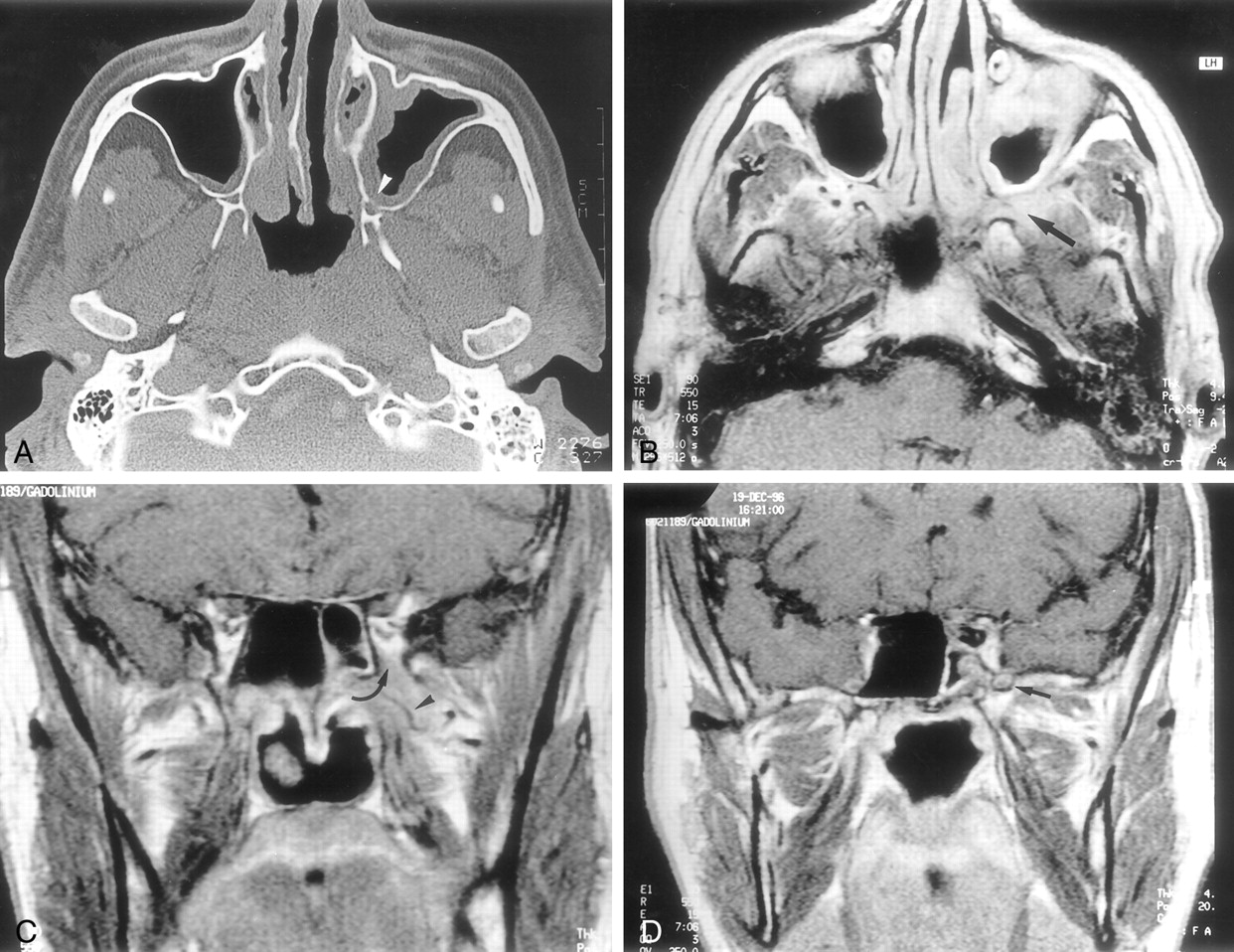
CT and MR imaging at presentation
A, Axial nonenhanced CT scan shows subtle bony lysis of the posterior wall of the left maxillary sinus (arrowhead). Note mucosal thickening of the left maxillary sinus.
B, Axial contrast-enhanced T1-weighted (550/15/3) MR image of the infratemporal fossa shows a poorly defined enhancing mass within the left PPF (arrow).
C, Coronal contrast-enhanced T1-weighted (550/15/3) MR image of the infratemporal fossa shows a soft-tissue mass of intermediate signal intensity (arrow), encasing the maxillary artery (arrowhead) and extending into the inferior orbital fissure.
D, Coronal contrast-enhanced T1-weighted (550/15/3) MR image, 8 mm posterior to C, shows enhancement of the left foramen rotundum (arrow).
Treatment was initiated with an intravenous bolus of methylprednisolone followed by oral prednisone, daily low-dose cyclophosphamide, and co-trimoxazole (trimethoprim-sulfamethoxazole). The trigeminal neuropathy resolved promptly. Follow-up CT 6 months later showed resolution of all previous abnormalities. After leukocytopenia occurred, cyclophosphamide was replaced by methotrexate for a period of 6 months. Long-term therapy then was pursued with low-dose prednisone and co-trimoxazole. Two years later, the patient remains in remission and CT and MR imaging follow-up findings confirm regression of anomalies (Fig 2).
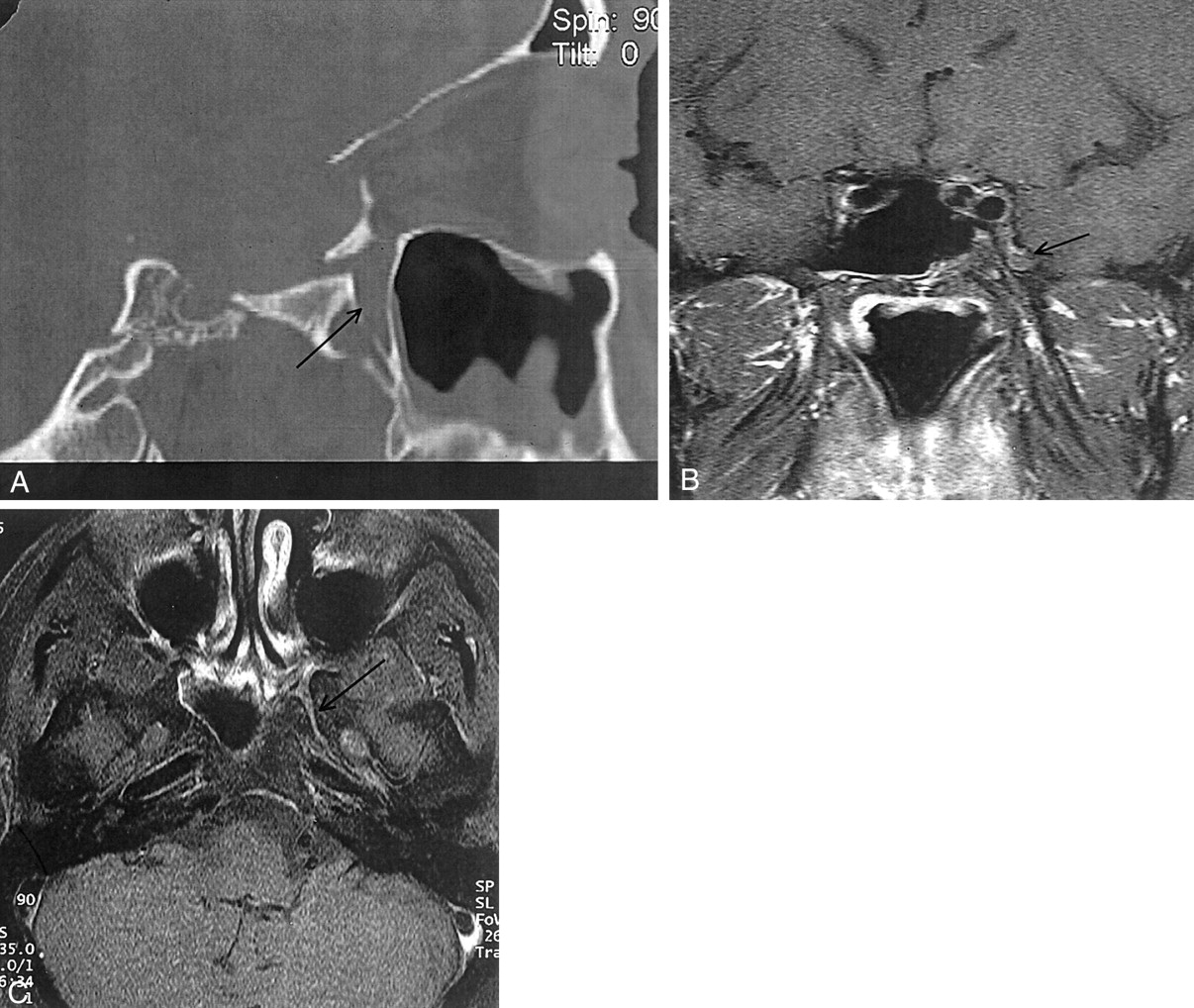
CT and MR images obtained 2 years after presentation.
A, Sagittal reconstructed high-resolution CT section. Note persistence of the mucosal thickening of the left maxillary sinus with regression of the palatine bony lysis (arrow).
B, Postcontrast T1-weighted image with fat saturation, coronal section. There is no enhancing soft-tissue mass involving the PPF or the inferior orbital fissure. Reduced signal intensity surrounds the pterygoid canal and the foramen rotundum (arrow).
C, Delayed (obtained 1 hour later) postcontrast T1-weighted image with fat saturation, axial section. Subtle enhancement of the PPF and foramen rotundum (arrow) exists compared with that of the normal contralateral side.
Discussion
WG is a systemic disease of unknown origin, characterized by a clinicopathologic complex of necrotizing granulomatous vasculitis of the upper and lower respiratory tracts and glomerulonephritis. Disseminated vasculitis may occur (1–3, 6). In addition to the upper and lower respiratory tract, this multisystem disorder can involve the orbit, ear, central and peripheral nervous systems, skin, heart, breast, salivary glands, gastrointestinal tract, spleen, urogenital tract, and bone (4, 7).
According to the 1990 classification by the American College of Rheumatology, WG can be distinguished from other forms of vasculitis if two of the four following criteria are present: nasal or oral inflammation, abnormal findings on chest radiographs, abnormal urinary sediment, or granulomatous inflammation at biopsy (8). Recently, a classification with three forms of WG was proposed. To the two classic forms (the generalized diffuse form and localized form without airway and renal involvement) has been added a purely granulomatous form (without evidence of vasculitis) involving primarily the ears, nose, and orbit. Each of these forms may be the only manifestation of WG. The locoregional form of the disease is common (≈30%) (9) and may last from months to years, but it may eventually progress to the classic generalized form.
The mean survival of an untreated, generalized form of WG is 5 months; 82% of patients die by 1 year (2), mainly from renal failure or secondary infection. Therefore, early diagnosis is important and can be achieved with a c-ANCA test (9, 10). This test has a high specificity—greater than 90% and as high as 99% if specific antiproteinase-3 autoantibodies are present (9)—and high sensitivity (60% in local-regional disease) (3, 9, 10). A single negative c-ANCA finding does not exclude WG, and several determinations may be required for the diagnosis of WG. An immunosuppressive therapeutic trial involving corticosteroids and cyclophosphamide should be conducted in patients with a positive c-ANCA result and a strong suspicion of WG, even in those with negative biopsy results (11).
Cranial nerve involvement is not rare with WG, but it has rarely been described as a sole presenting symptom (1). Neurologic involvement in WG, including cranial nerve palsy, peripheral neuropathy, cerebrovascular events, cerebritis, and miscellaneous involvement is a well-known phenomenon and occurs in 22–54% of patients (2, 6). In a recent study of 324 patients with WG, Nishino et al (1) found only 21 cases with cranial nerve involvement, seven with it as a presenting symptom, and five patients with trigeminal neuropathy (external ophthalmoplegia being classified separately). Drachman (6) classified three patterns of neurologic involvement: direct granulomatous involvement, remote granulomatous lesions, and systemic vasculitis. We postulate that the palsy of the palatine nerve observed in our patient may have been produced by direct encroachment of paranasal granuloma, resulting in hypesthesia of the mucosal surface of the hard palate. Direct granulomatous extension within the superior orbital fissure explains ocular proptosis. Moreover, the MR imaging pattern of involvement of the sphenopalatine canal and the foramen rotundum suggests granulomatous perineural spread as the origin of the trigeminal nerve palsy.
Perineural spread of tumor occurs with malignant and benign tumors of the head and neck as a form of metastatic disease in which tumor disseminates to noncontiguous regions along the endoneurium or perineurium (5, 12). The PPF is an important landmark, because it is a branching point for the trigeminal nerve. Perineural spread is a serious finding to recognize before any treatment. Pain, paresthesias, and denervation are frequent clinical findings that suggest the perineural spread of tumor. Therefore, in patients with cranial nerve abnormalities, radiologists should exclude abnormalities along the course of the affected nerve. The main features on CT or MR images include foraminal enlargement or destruction, fatty tissue obliteration, and nerve enlargement or enhancement. The imaging findings of perineural spread have rarely been described to occur with inflammatory lesions such as orbital pseudotumor (13), in actinomycosis (14), and in WG (11).
Patients with chronic rhinosinusitis associated with trigeminal neuropathy should undergo high-resolution CT examination, including bone and soft-tissue window-setting scanning. Because of its multiplanar capability and its high soft-tissue contrast enhancement, an MR imaging study of the entire cranial nerve V pathway should be performed. Axial and coronal scans of the foramen rotundum and the Meckel cave should be performed to detect a lesion of the trigeminal nerve. The use of contrast-enhanced T1-weighted images with fat suppression increases the sensitivity to depict perineural spread (2).
Our report shows that, as described (15), WG of the PPF may present as an inflammatory pseudotumor. We recommend that patients with a clinical and histologic diagnosis of pseudotumor of the PPF undergo c-ANCA serum assay. Cytotoxic therapy initiated after early diagnosis of WG gives the patient the best potential for remission. Cranial nerve involvement in WG may be explained by direct involvement of the nerves or by vasculitis.
Acknowledgments
The authors thank Walter Grauer, MD (Zurich, Switzerland) for his accurate advice and Daniel Rochet for the images.
Footnotes
Presented as an abstract at the 36th Annual Meeting of the American Society of Head and Neck Radiology, Phoenix, AZ, April 1998.
References
- Received October 5, 1999.
- Accepted after revision August 21, 2001.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.