
Abstract
Summary: We report a case of presumed vacuolating megalencephalic leukoencephalopathy (VML) in a 5-year-old boy that was diagnosed when the patient was 34 months old. The patient presented clinically with megalencephaly and delayed onset of slowly progressive neurologic dysfunction. Serial MR imaging and biochemical analysis were performed to differentiate VML from other disease entities with megalencephaly and leukoencephalopathy of infantile onset. Information obtained with diffusion tensor MR imaging revealed nearly intact white matter anisotropy and vectors in parietal and posterior temporal lobes in which hyperintense abnormality was shown on conventional T2-weighted images, and proton MR spectra showed a significant decrease in 𝓃-acetylaspartate-creatine + phosphocreatine ratio.
Vacuolating megalencephalic leukoencephalopathy (VML) (leukoencephalopathy, megalencephaly, and mild clinical course; van der Knaap disease) is a new entity of neurodegenerative disorders characterized by infantile-onset megalencephaly, cerebral leukoencephalopathy, and a delayed onset of slowly progressive neurologic dysfunction (1–8). In contrast to the relentlessly progressive infantile-onset leukoencephalopathy that is frequently fatal within the first decade of life, such as with Canavan disease, Alexander disease, and GM1 gangliosidosis, VML has the distinguishing feature of a remarkably slow course of deterioration in neurologic function. Neuropathologic appearance of VML is that of a spongioform leukoencephalopathy (3), although the cause of this leukoencephalopathy is still unknown. In light of the potential sensitivity of diffusion tensor (DT) MR imaging in the assessment of white matter diseases, as proposed by recent studies (9–11), we present the serial DT imaging and proton (1H) MR spectroscopy findings in a 5-year-old boy with VML.
Case Report
This 5-year-old boy presented to our department at 32 months old with progressive megalencephaly and delay in walking and speech. Progressive megalencephaly was noted before the patient was 1 year old. Head circumference was 49.8 cm when he was 14 months old, which was above the 97th percentile, and height and weight were within the 25th–50th percentile. Neurologic examination revealed normal muscle tone and tendon reflex and mild ankle clonus in bilateral lower legs. The patient was unable to walk independently until he was 21 months old and he could not say “papa” or “mama” until he was 36 months old. A mildly ataxic gait developed slowly after the patient was able to ambulate. He had one episode of febrile convulsion at age 17 months and subsequently developed myoclonus and complex partial seizures since the age of 3 years. At follow-up, he had a persistently large head circumference (>97th percentile) and mildly ataxic gait on examination at the age of 5 years 4 months.
Laboratory findings from routine serum and urine biochemical profile analysis, enzymatic studies for lysosomal diseases (metachromatic leukodystrophy), plasma amino acid analysis, organic acid urine chromatography (including 𝓃-acetylaspartate [NAA] and L-2-hydroxyglutarate), and plasma very long chain fatty acid study showed levels within normal limits. Neurophysiologic examinations showed normal motor and sensory nerve conduction velocities, brain stem auditory and visual evoked response, and somatosensory evoked potentials. Electroencephalography (EEG) at 17 months of age yielded normal findings, and follow-up EEG at 3 years of age showed intermittent 3–4-Hz slow waves located anteriorly and generalized paroxysmal bursts of 2–2.5-Hz spike-and-slow waves and frontal foci of spikes were also evident. Neurodevelopmental assessment by using the Wechsler Preschool and Primary Scale of Intelligence-Revised (WPPSI-R) was performed when the patient was 4 years old. The WPPSI-R full-scale intelligence quotient was 71, verbal subscale 71, and performance subscale 74. Borderline mental retardation was concluded.
MR examinations were performed with a 1.5-T system (Magnetom Vision; Siemens Medical Systems, Erlangen, Germany) equipped with a standard circularly polarized clinical radio-frequency head coil. In addition to the conventional T1-weighted, T2-weighted, and contrast-enhanced T1-weighted images obtained,1H multivoxel MR spectra and DT images were also acquired. Spectra were obtained with a point-resolved technique (TR/TE, 1500/135; field of view, 16 cm; phase encodings, 16 × 16; section thickness, 10-mm; and spectral width, 1 KHz). DT information was collected along six gradient directions by using a custom single-shot spin-echo echo-planar MR pulse sequence with Stejskal-Tanner gradients. The directional pattern used for the six gradients was at (x, y, z) = [(1, 1, 0), (1, −1, 0), (0, 1, 1), (0, −1, 1), (1, 0, 1), (−1, 0, 1)]. The approximate b value was 1000 s/mm2 for each of the six directions. An additional image was obtained with no diffusion sensitization (b = 0). DT image acquisition parameters were as follows: TR/TE, 4700/120; matrix size, 128 × 128; section thickness, 5-mm; and sections, 16.
Cranial MR imaging performed when the patient was 2 years 10 months old revealed diffuse T2 hyperintensities suggestive of demyelination involving mainly the fronto-temporo-parietal lobes and sparing the occipital subcortical regions (Fig 1A) and frontal periventricular linear hyperintensities on T1-weighted images (Fig 1B). The bilateral globus pallidi showed a slight increase in signal intensity on T1-weighted images and slightly enhanced with intravenous injection of 0.1 mmol/kg gadolinium dimeglumine (Fig 1C). There was no subcortical cyst observed on the MR images. Follow-up MR imaging at age 5 years showed essentially unchanged cerebral white matter abnormalities but a larger cavum septi pellucidi without the presence of subcortical cysts (Fig 2).

MR images obtained when the patient was 2 years 10 months old.
A, Axial fast spin-echo T2-weighted image (TR/TE, 3500/93; two signals acquired) at the level of the basal ganglia, showing diffuse demyelination involving mainly the frontal lobe sparing the occipital subcortical regions and internal capsule.
B, Coronal T1-weighted contrast-enhanced image (700/14), showing periventricular linear enhancement next to bilateral frontal horns.
C, Axial T1-weighted contrast-enhanced image (700/14), demonstrating enhanced lesions in bilateral globus pallidi (arrows).
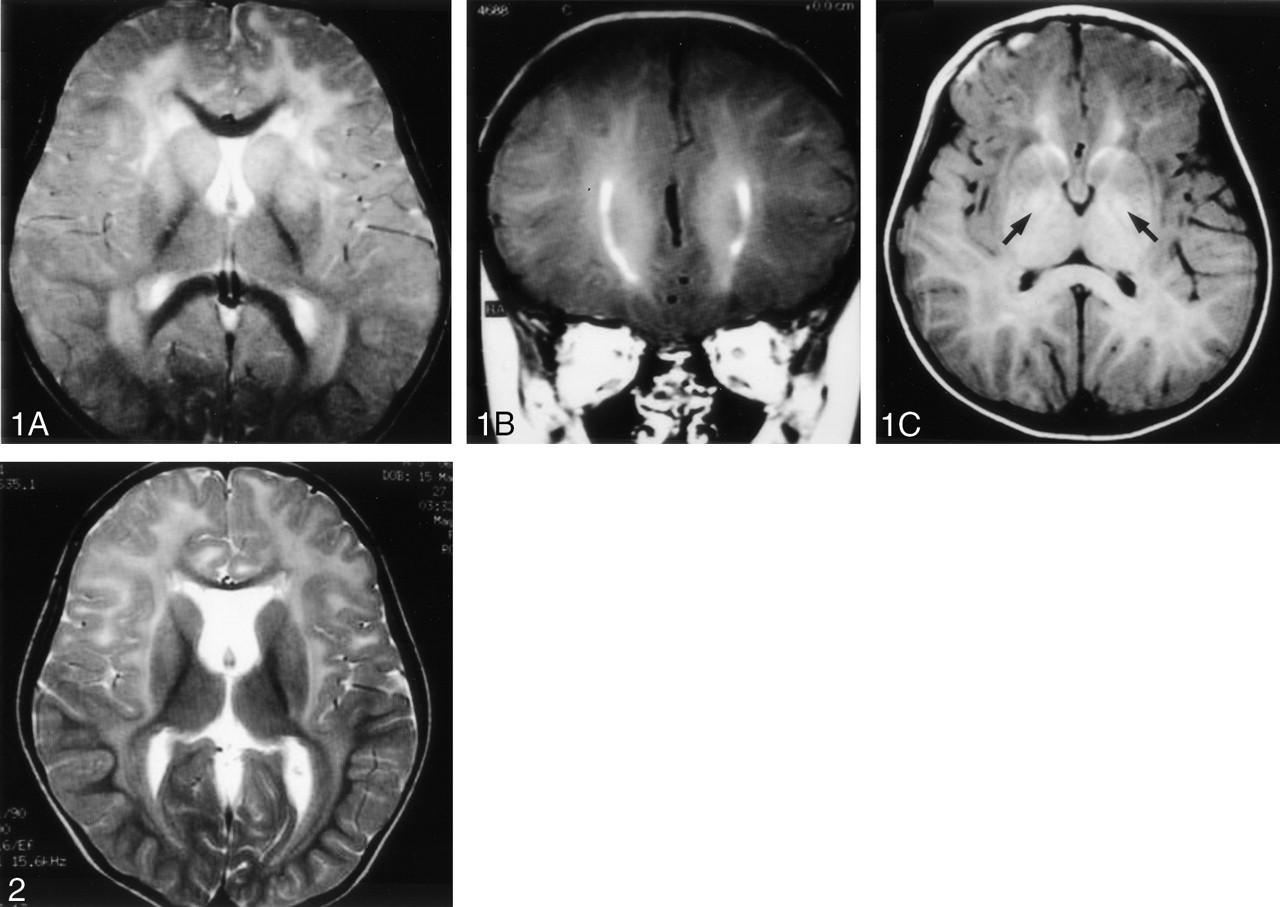
Follow-up axial T2-weighted MR image (4000/97.6), obtained when the patient was 5 years old, at the level of the basal ganglia, showing consistent demyelination at the same regions on Fig 1A with thinning of the genu of the corpus callosum and dilatation of the cavum septi pellucidi.
On the DT images, the vector map and fractional anisotropy (FA) map showed severe loss of anisotropic structure in frontal white matter area, whereas only mild abnormality was shown in parietal, posterior temporal, and occipital white matter areas by visual observation (Fig 3). To evaluate the integrity of anisotropic structures, a quantitative comparison of FAs was made between the patient with VML (n = 1) and age-matched control subjects (n = 2). The regions of interest placed on regions with high-signal-intensity abnormalities seen on T2-weighted images were manually drawn on FA maps as shown in Figure 4. The recorded values for FA within each region of interest were averaged with contralateral homologous region-of-interest values. The Table lists the FA from the patient with VML and the corresponding data from the controls for each region of interest. White matter anisotropies were largely reserved, and FA was decreased from 7.1% to 23.8%. Multivoxel1H MR spectra showed a decreased NAA-creatine (Cr) + phosphocreatine ratio, elevated choline, and lactate peaks in bilateral temporo-parietal white matter regions (Fig 5).

MR and DT images obtained when the patient was 5 years 8 months old.
Axial T2-weighted MR image (A) at the level just above the body of the corpus callosum, showing diffuse high-signal-intensity white matter abnormalities. On FA (B and D) and vector maps (96 × 128 matrix size) (C and E), the white matter anisotropy and directionality were only mildly impaired (arrows) in our patient (B and C) as compared with a healthy control subject (D and E).

Depiction of regions of interest used in data analysis obtained when the patient was 5 years old. Each region of interest was placed symmetrically and bilaterally except for region of interest 4.

Multivoxel 1H MR spectra, obtained when the patient was 5 years old showing a significant decrease of NAA/Cr + phosphocreatine. Left spectrum, which is a magnified voxel indicated with thick frame in multivoxel 1H MR spectroscopy, shows the presence of lactate peaks in the temporoparietal white matter regions.
Mean FA of a patient with vacuolating megalencephalic leukoencephalopathy and of control subjects
Discussion
VML is a rare neurodegenerative disorder characterized by infantile-onset megalencephaly, cerebral leukoencephalopathy, and delayed onset of slowly progressive neurologic dysfunction. The megalencephaly typically develops during the 1st year of life. Initial mental capacities are normal, and motor development is mildly delayed in most patients. Subsequently, slowly progressive ataxia and spasticity develops, although intellectual functioning is preserved for years after onset of the disorder (3, 4). About 25–75% of patients with VML develop epilepsy that usually responds well to anticonvulsant therapy (1, 5). Characteristic subcortical white matter lesions can be seen with MR imaging, but MR-depicted lesions do not reflect the severity of this disease (5). Autosomal recessive inheritance has been assumed to be partly attributed to the high rate (56.5%) of consanguinity among the parents and presence of affected siblings (1, 3, 5). The case herein presented with the typical slowly progressive course, which, to our knowledge, has not been seen in other types of infantile-onset leukoencephalopathy.
Histopathologic study has shown VML to be one of the vacuolating myelinopathies; that concurs with MR imaging evidence of a spongiform leukoencephalopathy without cortical involvement (3). Light microscopy shows intact, myelinated fibers of normal or nearly normal density and staining properties. Electron microscopy shows myriad vacuoles in subcortical white matter, most of which are covered by single, five-layered membranes representing single myelin lamellae. These vacuoles are concentrated in the outermost lamellae of myelin sheaths, leaving the remainder of myelin sheaths undisturbed. Despite severe myelin vacuolation, myelin paucity and the axonal damage are not noted. These features may be related to the mildly progressive course of neurologic dysfunction in the patients with VML (3).
Previous reports have shown that the characteristic MR findings of VML were diffusely abnormal in signal intensity and swelling of the cerebral white matter with cystlike spaces in the frontoparietal and anterior temporal subcortical areas as well as sparing of periventricular rim of occipital white matter, basal ganglia, corpus callosum, and the anterior limb of internal capsule (1). The cyst tends to be bilateral and enlarges over time; cysts increase in number with advancing age (4). In all reported cases, the cavum septi pellucidi persisted as did the cavum vergae in some patients (1, 5). Topcu et al (5) proposed a visible claustrum sign in which the sparing claustrum was clearly visualized in contrast to the diffusely demyelinated external and extreme capsules on T2-weighted images. Increased T2 signal intensity was also found at the interface of the putamen and globus pallidus in one patient. Our MR findings are consistent with those reported previously, except for the periventricular linear hyperintensities next to bilateral frontal horns seen on unenhanced T1-weighted images and the absence of the subcortical cyst. The high-signal-intensity T1 lesions may be attributed to ischemic gliosis, microcalcifications, or aggregations of T1-shortening substances, including free radicals, in the deep white matter regions. These T1 high-signal-intensity changes may sometimes be mistaken for lesions caused by other leukodystrophy such as those in Alexander disease, in which the presence of frontal periventricular white matter enhancement after gadolinium dimeglumine administration is considered an important imaging sign.
In addition to conventional MR imaging, we also employed DT imaging, a technique that depicts microscopic water diffusion anisotropy in normal and abnormal white matter. Previous investigations have shown white matter anisotropy loss in disorders such as multiple sclerosis, age-related leukoaraiosis, and leukoencephalopathy (9). Anisotropy maps and indices are sensitive to subtle anisotropy loss in patients with Krabbe disease; T2-weighted imaging cannot depict this abnormality (10). In patients with adrenoleukodystrophy, Melhem et al (11) also reported different degrees of anisotropy loss in regions of abnormal T2-weighted hyperintensity, which may help differentiate between potentially reversible and irreversible damage in the white matter. Our results of the vector and FA map showed severe loss of anisotropy in the frontal white matter area, whereas only mild abnormality was shown in the parietal, posterior temporal, and occipital white matter areas; no changes of major diffusion directionality were noted. Because FA indices may reflect the microstructural property of the white matter, the mild alternation of diffusion anisotropy in our case might suggest, to some degree, the structural integrity of the white matter in these areas. As shown histopathologically, in VML only the outermost myelin lamella remain uncompacted and converted into large vacuoles between the structurally intact myelin sheath (3). The relatively preserved myelin sheath might also account for the nearly normal FA indices. In contrast, significant reductions in FA indicate loss of an ordered structure governing the directionality of water molecule displacement; this can serve as a marker for permanent white matter injury, such as that of frontal white matter in our case. On the basis of these results, we posit that findings of anisotropy maps may correlate with neurologic function and may explain the slowly progressive course of neurologic dysfunction in the present case.
VML can be distinguished from other disease entities with megalencephaly and leukoencephalopathy of infantile onset such as Alexander disease, Canavan disease, L-2-hydroxyglutaric aciduria, and one variant of congenital muscular dystrophy (1) on the basis of clinical, neuroimaging, and biochemical findings. For example, rapid clinical deterioration that leads to severe disability and early death within a few years in infantile Alexander disease was not found in our patient (7). Features of Canavan disease, including elevated NAA in urine and brain, including involvement of thalamus and globus pallidus (1, 5, 7), also were not found in our case. MR imaging of L-2-hydroxyglutaric aciduria that typically shows involvement of caudate nuclei, putamen, and dentate nuclei, together with severe external hydrocephalus and leukoencephalopathy, was not observed in the present case. Childhood ataxia with diffuse cerebral hypomyelination syndrome can be distinguished from our patient by the absence of macrocephaly in the 1st year of life. The diagnosis of VML in the present case was determined from clinical and MR imaging findings, and other diagnoses were excluded, despite the absence of a subcortical cyst. It is clear that age and stage dependencies must be taken into account and that follow-up studies during disease progression are warranted.
It has been reported that no basic biochemical defect was identified in patients with VML (1–3, 5). Nonetheless de Stefano et al’s (12)1H MR spectroscopy study involving two patients showed a significant decrease in NAA-Cr + phosphocreatine ratio and concomitant small increases in lactate in the white matter of both hemispheres. MR spectroscopy findings in the case herein support their results; the metabolic profile may continue to deteriorate despite no evidence of definite metabolic defect evinced with biochemical analysis. NAA is believed to be of neuronal and axonal origin in mature brain, and its decrease is most likely due to axonal loss or dysfunction. Low NAA may be the first sign of metabolic impairment in patients with VML. Choline increase is most likely due to enhanced myelin turnover related to demyelination and accumulation of membrane myelin degradation products. On the basis of our DT and1H MR spectroscopy findings, we theorize that axonal loss and cell accumulation associated with early-onset demyelination may not have a prominent effect on diffusional properties in ordered axonal systems.
By use of clinical and MR imaging findings, we demonstrated that in presumed VML cases the white matter anisotropy may be shown to be reserved at DT imaging and the NAA-Creatine + phosphocreatine level decreased despite disease progress and the absence of biochemical defects evinced at laboratory investigations.
References
- Received May 15, 2003.
- Accepted after revision November 18, 2003.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.