
Abstract
Summary: Degos disease, or malignant atrophic papulosis, is a rare obstructive vasculopathy of unknown origin, characterized by distinctive skin lesions, visceral involvement, and an unfavorable outcome. The gastrointestinal tract and the central nervous system are most frequently affected, but cases limited to benign skin lesions have also been described. Neuroradiologic reports of this condition are exceptionally rare. We report the case of a 29-year-old woman with central and peripheral nervous system involvement who presented with progressive clinical deterioration and a meningovascular pattern at cerebral MR imaging; other organs were spared in this patient.
Degos disease (also known as malignant atrophic papulosis or Kohlmeier-Degos disease) is a rare vasculopathy of unknown origin, characterized by vascular lesions of the skin, gastrointestinal tract, and CNS (1, 2); a genetic predisposition and an immunologic pathogenesis are suspected.
Distinctive cutaneous lesions (erythematous papules with “porcelain” white center) are the first signs of the disease, which are followed by a variable, progressive involvement of visceral organs. Heart, eyes, lungs, and kidneys are also involved. The onset of a systemic diffusion of the disease leads to death in several months or years, the most frequent fatal complication being intestinal hemorrhage and perforation. Despite the term “malignant” being present in its name, there have been several reports of cases with only cutaneous manifestations and a favorable prognosis (3, 4).
The neurologic manifestations of Degos disease include cerebral infarcts, subdural hematomas, venous sinus thrombosis, polyradiculoneuropathy, and nonspecific symptoms without objective findings. Less frequently, myelopathy and myopathy are described (4). We describe a patient with neurologic manifestations, characterized by a severe involvement of the small vessels and the cerebral meninges.
Case Report
A 29-year-old woman, with a family history of hypertension, reported the onset of her symptoms at approximately 26 years of age, when skin lesions appeared over her upper and lower extremities. Such lesions were few and isolated at the beginning but extended to the whole body in a few months and were accompanied by intense fatigue, paresthesia, diplopia, ataxic gait, and sphincter function abnormalities. Several hospital admissions followed, and, after an initial diagnosis of “primary vasculitis,” the typical skin lesions of Degos disease were recognized. Clinical tests and laboratory data showed a central and peripheral nervous system involvement, but various therapeutic attempts with antiinflammatory drugs, immunosuppressant treatments, and plasmapheresis resulted in no substantial clinical improvement.
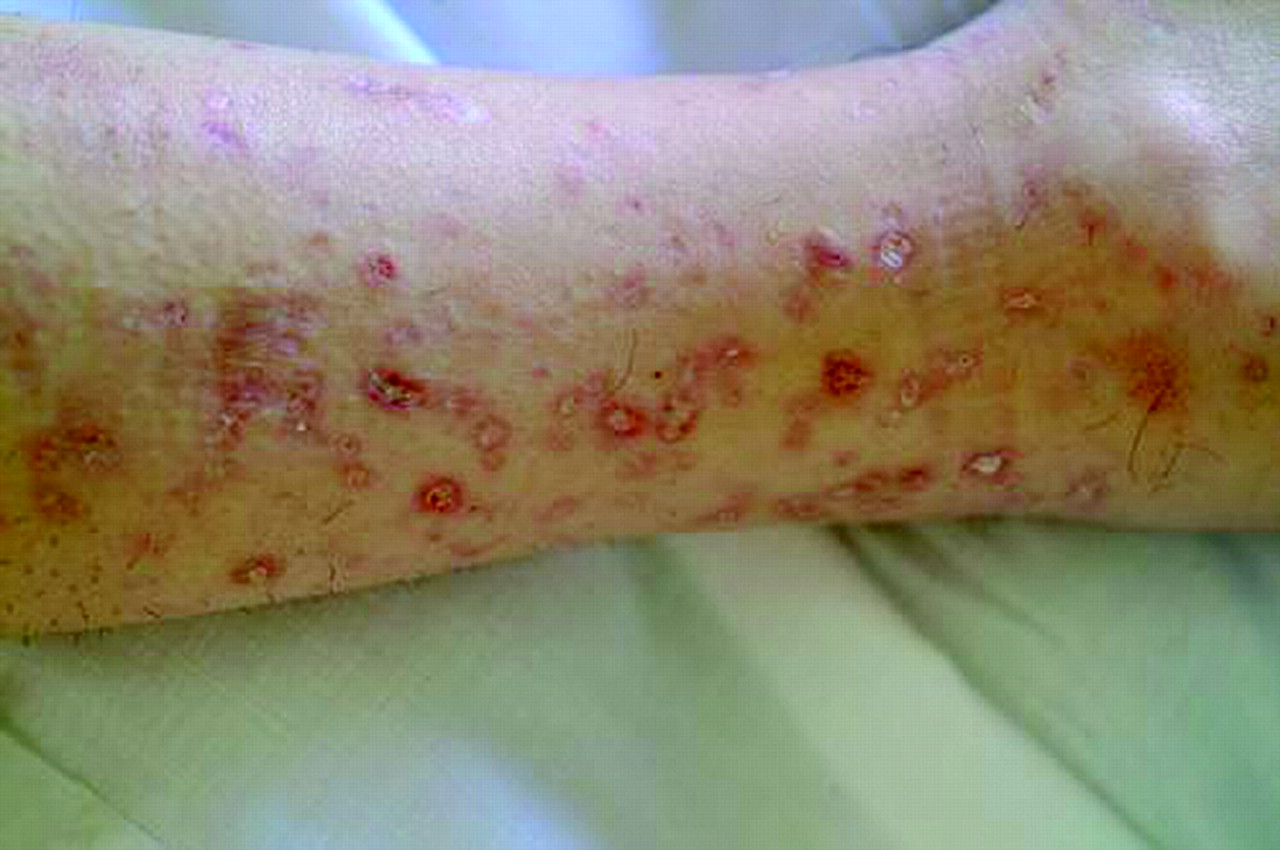
On the dermatologic examination, diffuse asymptomatic papules were found, mostly in the lower limbs. These lesions had a diameter of a few millimeters, were surrounded by a erythematous rim, and their color was pink, with a central atrophic area characterized by adherent whitish desquamation (Fig 1); a skin biopsy on the leg confirmed the diagnosis of Degos disease (Fig 2). The neurologic examination noted anisocoria, hearing loss on the left side, diffuse muscle hypotrophy, impaired finger movements, mild paresis of the right arm, paralysis of both legs, symmetrically reduced muscular reflexes, hypoesthesia, hypopallesthesia of the lower limbs, constipation, and urinary incontinence. No fever or meningeal signs were present. Brain MR imaging (Figs 3–5) showed both old and new ischemic lesions, mainly present in the left hemisphere, and numerous small nodules, localized in both the cerebral and the cerebellar cortices. A diffuse and homogeneous thickening of the meninges, with some subdural fluid accumulation, was also evident in the frontoparietal region. Ependymal enhancement was noted in the atrium of the right lateral ventricle. Hydrocephalus and venous thromboses were ruled out. Spinal MR imaging showed thinning of the spinal cord, with mild alteration of the signal intensity in the thoracic cord (Fig 6). A subtle, focal cord enhancement was found in the cervical and thoracic cord (Fig 7).

Typical skin lesions of Degos disease. Papules on patient leg, at different stages, with whitish, atrophic center and erythematous rim.
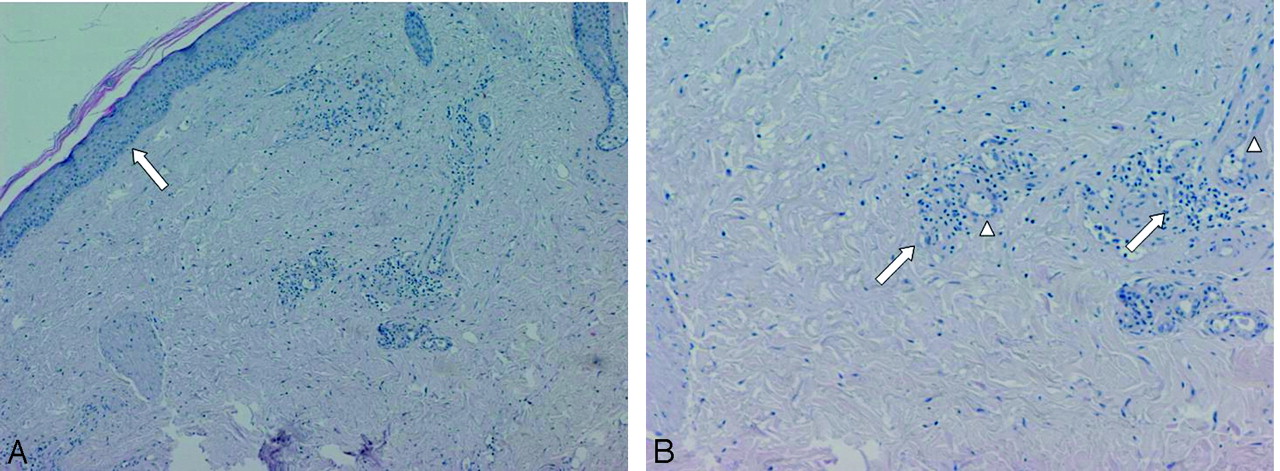
Skin histologic specimen showing atrophic epidermis with no crests (A, arrow) and low cellularity and focal necrosis in underlying dermis (Hematoxylin-eosin). High magnification of deep dermis (B) revealing arteriolar hyalinosis with endothelial hyperplasia (arrowheads) and perivascular lymphomonocitic infiltrate (arrows).

Contrast-enhanced T1 axial images show multiple nodular lesions located in the peripheral gray, subarachnoid spaces and leptomeninges, with diffuse dural enhancement. Note the ependymal enhancement (A) of the right atrium and a small infarct in the left caudate head.
The different blood tests performed showed only a fibrinogen level of 750 mg/100 mL. No relevant thoracic and abdominal findings were found. The patient was discharged home with a symptomatic therapy composed of antiplatelets and antidepressant drugs and the indication of a neurologic rehabilitation program.
Discussion
Degos disease generally affects young adults and is characterized, at the beginning, by typical skin lesions with papulous aspect, erythematous rim, and white porcelainlike atrophic center, localized over the trunk and the lower limbs. These lesions are asymptomatic or mildly itchy (5, 6). After a variable period of weeks, months, or (in some cases) years, the pathologic process involves also other organs and, in particular, the gastrointestinal tract (50% of cases) and the CNS (20–60% of cases) (4). Eyes, urinary tract, heart, and lungs can also be involved, but with a lower incidence (7, 8). The multisystem involvement is often fatal and generally leads to death in 2–3 years, most frequently as a consequence of intestinal perforation (6). Beyond this classic presentation, many cases with favorable evolution and good prognosis have also been reported in recent years, which showed a prevalent or exclusive involvement of the skin (9). In particular, in a large series from the Mayo Clinic more than half of the patients have been reported as almost asymptomatic (4), and therefore this disease should be reclassified to reflect the existence of a benign and a malignant form.
Several reports of familial forms of the Degos disease support the hypothesis of a genetic predisposition for this condition (10). The pathogenetic mechanism most frequently indicated in the literature is immunologic, because abnormal plasma levels of antibodies have been found (antiphospholipids and, particularly, anticardiolipins), along with deposits of immunoglobulins in the derma and epidermis of the skin lesions (9, 11, 12). Autoimmune phenomena have been involved in such findings, which might cause first endothelial damage and, subsequently, abnormal activation of the coagulation system (13).
Histologic characteristics in both skin and systemic lesions include concentric thrombogenic thickening of the intima of small and middle vessels, caused by the swelling of the endothelial cells and the proliferation of the underlying fibromuscular cells. The presence of subendothelial lipid inclusions is thought to contribute to the thickening of the intima. The tunica media is usually not involved in the process. At the level of the adventitia, it is possible to find inflammatory-like perivascular infiltration, without typical histologic signs of vasculitis, such as those found in nodous polyarteritis and thromboangioitits (4, 13).
Skin biopsy of our patient revealed atrophic epidermis with no crests; underlying dermis showed low cellularity and focal necrosis (Fig 2A). In deep dermis arteriolar ialinosis, endothelial hyperplasia, and perivascular lymphomonocitic infiltrate were recognized (Fig 2B).
In the CNS, the most common abnormalities are a stenosis or obstruction of the small and middle subarachnoid and extraparenchymal arteries. Carotid, cerebral, and vertebrobasilar arteries usually show small fibrous subendothelial plaques; this apparent resistance to the pathologic process is probably due to different antigenic properties of the endothelium of such vessels (13). Veins can be secondarily involved by obstructive phenomena, when a nonspecific coagulative disorder might modify the activity of fibrinogen or platelet adhesion (14).
Fibrosis of the meninges, caused by collagen accumulation in the cortical sulci and in the subarachnoid spaces, constantly accompanies the vessel abnormalities and can be explained by the same mechanism of endothelial damage mentioned above. This might be the consequence of an autoimmune injury of arachnoid fibroblasts and mesothelial cells followed by reactive fibrosis or might be caused by a dysfunction of coagulation factors (13). Histopathologic characteristics of the spinal cord and of the peripheral nervous system have rarely been described and diffuse myelomalacia was discovered at autopsy in only one case (7).
In our patient, the most important radiologic aspect was the presence of disseminated small cortical nodules, expression of superficial vasculitis, and leptomeningeal fibroinflammatory deposits. The signs of ependymitis we found in the right lateral ventricle (Figs 3 and 4) seem to indicate that inflammatory phenomena were superimposed to the vascular damage, as already described in some forms of Degos diseases with a meningoencephalitislike presentation (15). In addition, the dural thickening, in the absence of venous thrombosis, is compatible with the hypothesis of a superimposed inflammatory process and represents a chronic fibroinflammatory finding. The presence of a small subdural fluid collection should be the result of a CSF equivalent effusion or of blocked CSF spaces rather than a chronic subdural hematoma (Figs 4 and 5). Finally, the involvement of the spinal cord was confirmed by the history and the clinical findings of our patient; we noted, beyond a global reduction in the spinal cord thickness, the presence of focal abnormalities in cervical and thoracic cord (Figs 6 and 7).

Contrast-enhanced FLAIR coronal image shows cerebral and cerebellar lesions, right ependymal alteration, and dural thickening; ischemic changes can be noted in the left parietal white matter. On the right, subtle hypointense CSF collection is recognizable among thickened meninges.

T2 (A) and T1 (B, contrast-enhanced) axial images at supraventricular level show right thickened meninges, subdural CSF collection (right side more evident than left) and multiple superficial lesions.
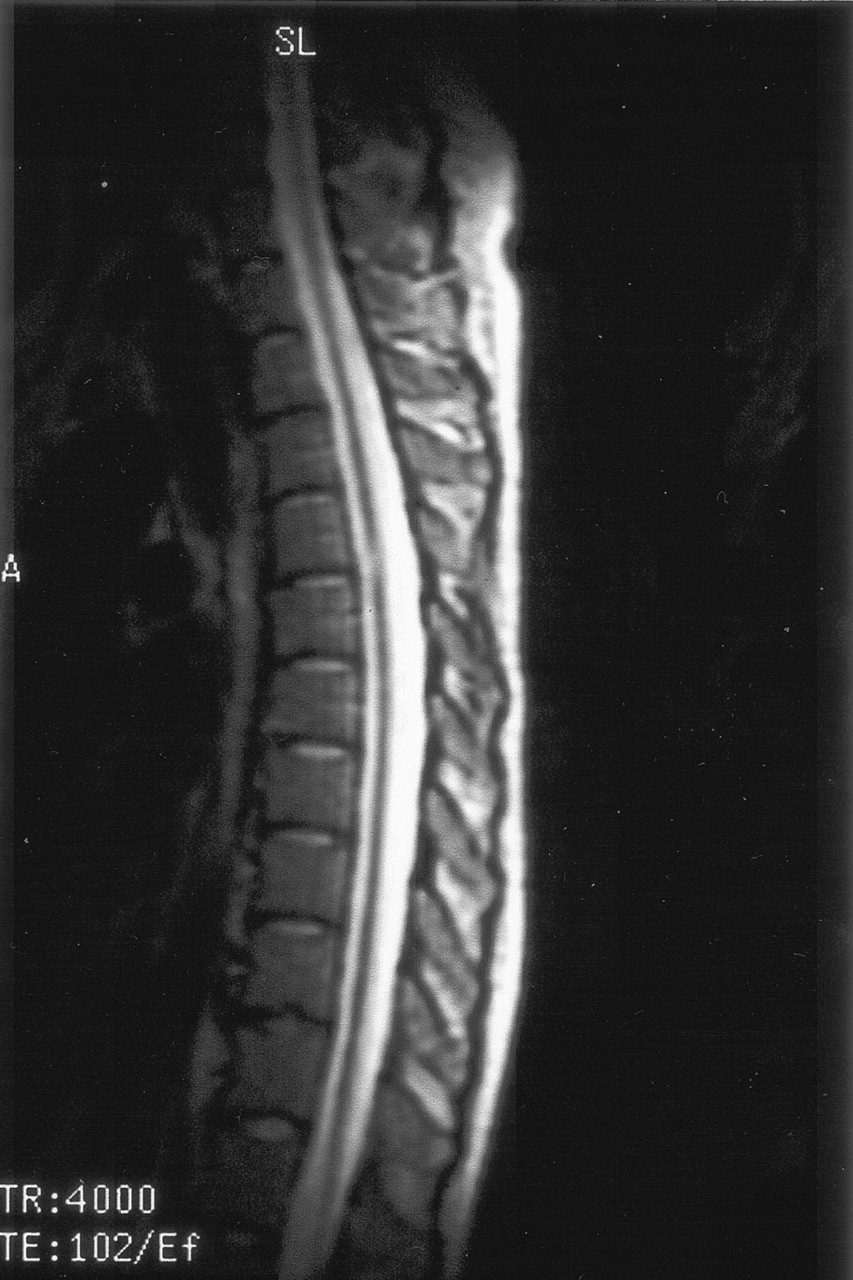
T2 sagittal image shows diffuse spinal cord thinning and midthoracic focal hyperintensity.
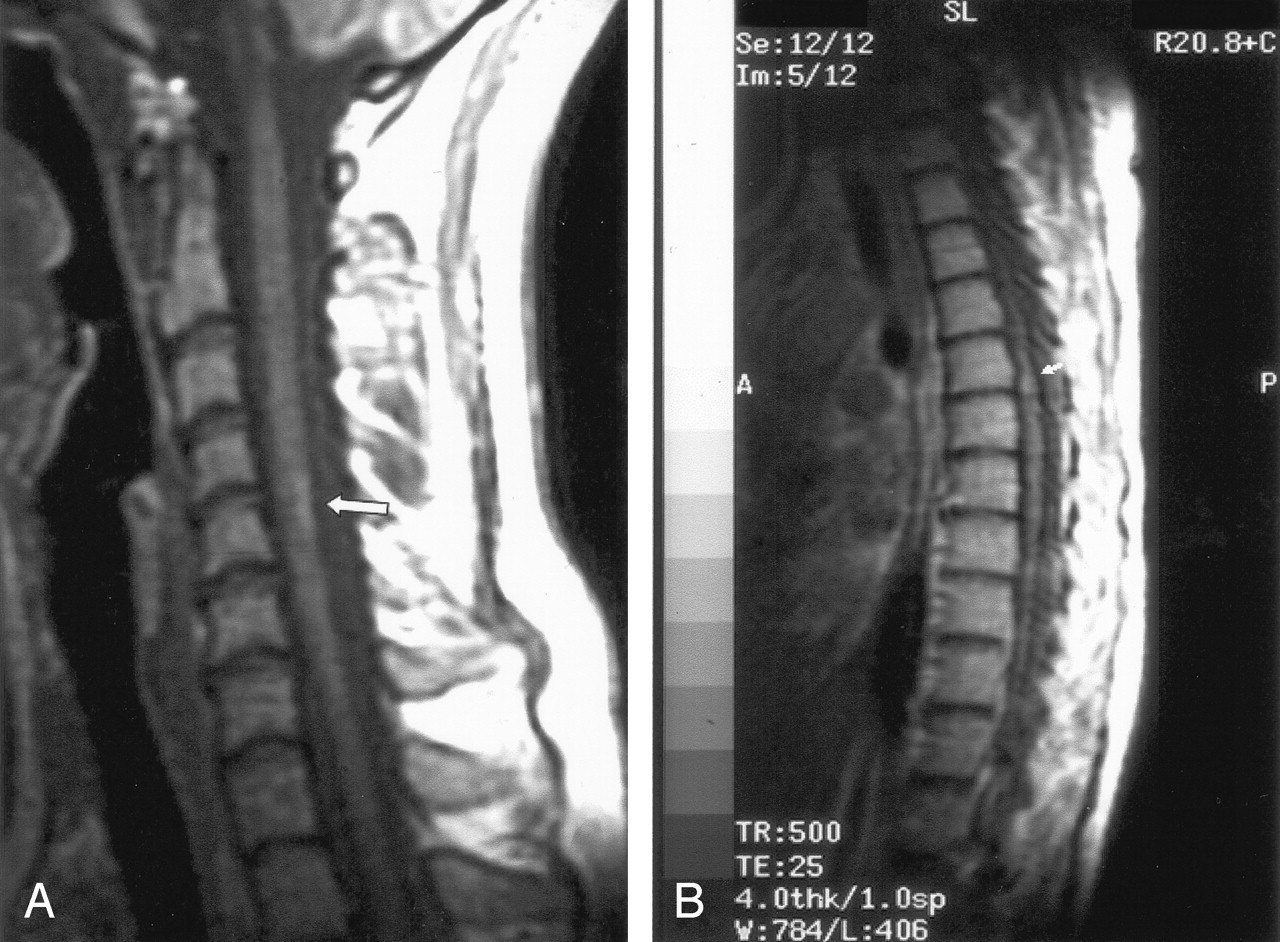
T1 (contrast-enhanced) sagittal images show subtle cord enhancement in the dorsal cervical region and at midthoracic level (arrows).
Conclusion
Our patient presented mainly with neurologic findings but without other systemic disorders, which are frequently reported for this disease in the literature. Moreover, it is likely that, in addition to the brain lesions and the spinal cord abnormalities, also the diffuse sensory-motor polyneuropathy, which was drug-resistant and already present at the early stages of the disease, played an important role in determining the serious functional problems characterizing the clinical evolution of this case.
References
- Received February 6, 2004.
- Accepted after revision June 21, 2004.
- Copyright © American Society of Neuroradiology









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}