
Abstract
SUMMARY: Association of leukoencephalopathy, cerebral calcifications, and cysts (LCC) is a rare disorder that was recently described. To our knowledge, only 2 reports, including 3 patients in each, have been published in the literature to date. Herein, we report a 19-year-old man with LCC who had neurological symptoms beginning in late adolescence. Clinically, he had rare convulsive seizures, slowly progressive pyramidal symptoms, and normal intelligence. On radiological examination, there were progressive calcifications in the basal nuclei and the cerebral white matter, as well as parenchymal cysts and diffuse abnormal signals of the white matter on T2-weighted sequences on MR imaging. On histopathological examination, angiomatous changes and secondary gliosis were demonstrated.
The association of leukoencephalopathy, cerebral calcification, and cysts (LCC) is a very rare entity, which was originally described in 3 patients by Labrune et al 1996.1 Following this original report, Nagae-Poetscher et al reported on an additional 3 patients in 2004.2 The clinical presentation of this entity includes decline of cognitive performance, rare convulsive seizures, and a mixture of extrapyramidal, cerebellar, and pyramidal signs.1,2 Progressive calcifications in the basal and cerebellar gray nuclei and the central white matter are found on CT. MR imaging reveals diffuse abnormal signals of the white matter on T2-weighted sequences.1,2 A special feature is the development of parenchymal cysts in the supratentorial compartment and cerebellum, leading to compressive complications and surgical considerations.1,2 Histopathologic examination reveals angiomatous-like rearrangements of the microvessels, together with degenerative secondary changes in other cellular elements.1
In this report, a new patient with LCC is presented, and clinical, neuroradiologic, and histopathologic findings of this rare and recently described entity are discussed in view of the relevant literature.
Case Report
A 19-year-old man was admitted to our hospital for the evaluation of generalized tonic-clonic seizures 4 years ago. He had experienced these convulsive seizures for the past 2 years. He was born after an uneventful pregnancy. His apparently healthy parents were not consanguineous. No abnormality was found on general physical, ophthalmologic, and neurologic examinations. Complete blood count, sedimentation rate, liver and renal function tests, serum thyroid and parathyroid hormones, calcium, phosphate, alkaline phosphatase, and lactate levels were within normal limits. Serological tests for cytomegalovirus, toxoplasma gondii, toxacara canis and cati, hydatic cyst, and HIV 1 and 2 were all negative. Cervical, thoracic, and abdominal CT examinations showed no abnormality. Scalp EEG showed minimal slowing in background activity at the left frontotemporal area with no epileptiform activity.
The initial CT showed numerous foci of calcifications scattered in the pons, basal ganglia, and all lobes of the brain. The supratentorial lesions were predominantly located in the subcortical white matter. Some of the calcified lesions were associated with cystic formations (Fig 1A). The diameters of the calcifications, together with the cysts, ranged from a few to 15 mm. Additional findings included ring enhancement of the cyst walls and decreased attenuation of the white matter surrounding some of the lesions. MR imaging showed more cysts associated with the calcifications than seen on CT, whereas the calcifications were appreciated less conspicuously on MR imaging. Abnormally increased signal intensity was noted in the bulbous, pons, bilateral cerebellar, and cerebral peduncles, both basal ganglia, as well as in both hemispheric white matter, whereas the cortical gray matter was spared. These high-signal-intensity areas were always surrounding a cyst and/or a focus of calcification with evident mass effect leading effacement of adjacent sulci (Fig 1B, -C). These features suggested that the high-signal-intensity areas represent vasogenic edema affecting the parenchyma. The intensity of the cysts was higher than that of the CSF on T2-weighted, T1-weighted and fluid-attenuated inversion recovery images. Contrast-enhanced scans showed ring enhancement in all cysts.
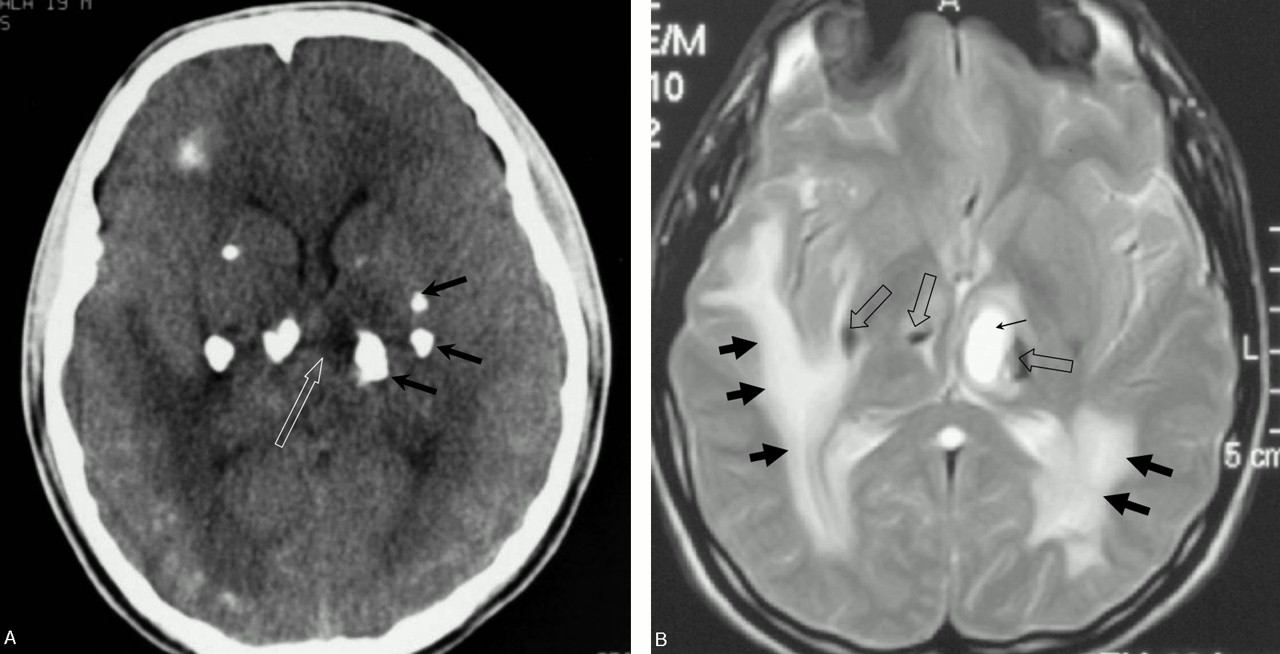

A, Initial CT scan through the basal ganglia shows a cyst (white arrow) and foci of calcifications (black arrows). Note also hypodensity of the white matter.
B, T2-weighted MR image through the basal ganglia shows the cysts (thin arrow), calcifications (hollow arrows), and the surrounding edema (thick arrows).
C, T2-weighted MR image through the cerebral peduncles shows the cysts (thin arrow) and the surrounding edema (thick arrows).
A diagnostic right temporal lesionectomy was performed. Histopathologic examination revealed edematous parenchyma with moderate gliosis and prominent angiomatous changes with microcalcifications (Fig 2A). Angiomatous changes were the numerous small, tortuous blood vessels, either contiguous or separated by nervous tissue, irregular, homogenous, periodic acid Schiff–positive thickening of the wall (Fig 2B). Staining red with Masson trichrome and negative reaction with Kongo red confirmed hyaline vascular thickening. Silver impregnation, as an indirect indicator of the elastic fiber degeneration, revealed fragmented rough reticulin fibers on the wall of most of the vessels. CD34 immunostaining showed regular, normal endothelial lining. Calcifications, mostly irregular and laminated, were all related to the abnormal vascular walls. Probable parasitic etiology was eliminated by negative reaction of the decalcified slides with methanamine silver. Foci of microhemorrhagia, ferric iron deposits (Prussian-blue positive), whirled, irregular Rosenthal fibers (reactive with glial fibrillary acidic protein immunostaining) mostly surrounding the abnormal vascular configurations were also noticed.

A, Small, tortuous, hamartomatous vessels with irregular calcifications (H&E, 100).
B, Numerous small, tortuous blood vessels with irregular, homogenous PAS positive thickening of the walls.
Valproic acid (VPA) 1000 mg/day was given orally to prevent seizures, and the patient was discharged from the hospital without symptoms and neurologic signs.
One year later, the patient was readmitted to our clinic because of repeated generalized tonic-clonic seizures and headache. On neurologic examination, he had mild hemiparesia and hyperreflexia on the left side. Measured serum level of VPA was 45 μg/dL, and the daily dose was increased to 1500 mg/day. Repeated CT and MR imaging examinations disclosed progression of the lesions. Diffusion-weighted imaging (DWI) and 1H-MR spectroscopy were also performed at this time, and DWI showed increased diffusion in the cysts. 1H-MR spectroscopy of the cysts showed only lactate peak but no evidence for the normal metabolites of the brain (Fig 3). During the 2-year follow-upperiod, some cysts enlarged gradually and caused compression to nearby structures, such as the ventricles, pons, and basal ganglia. When the patient was 21 years of age, the pontine cyst was surgically removed because of severe compression on the pons. A sample from the cyst wall was examined histopathologically. Rosenthal fibers and reactive gliosis were noted in the wall of the cyst.

Region of interest placed in the pontine cyst during 1H-MR spectroscopy. The spectrum from the cyst shows a doublet at 1.3 ppm representing lactate. (For the spectroscopy, TR = 1500 ms, TE = 144 ms.)
At the latest control, no additional findings were identified at neurologic examinations and the patient remained seizure-free; however, control CT and MR imaging showed further enlargement of some cysts, calcifications, and leukoencephalopathic areas.
Discussion
Although the presence of cysts, calcification, and perifocal white matter edema in our patient suggests a parasitic infection3—namely, cysticercosis or echinococcus alveolaris infection—no serologic or histopathologic confirmatory evidence was found. Regarding the relevant literature, LCC was considered. LCC is characterized by extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts.1 The onset may occur from early infancy to adolescence with signs including cognitive decline, convulsive seizure, and pyramidal, extrapyramidal, and/or cerebellar signs.1,2 Our patient experienced his first generalized seizure at 17 years of age. His seizures were rare and responded well to anticonvulsant drug therapy. The pyramidal symptoms developed 3years after the first convulsive seizure. Six years later, he had only still mild hemiparesia and hyperreflexia on left side. This slow evolution of the disease in our patient is in line with the very slow progression rate described for LCC.
In contrast to the mild clinical course, a severe and progressive neurodegenerative process was shown in the central nervous system on neuroimaging. CT and MR imaging findings in our case were similar to those of other cases described in the literature showing increased white matter signal intensity relatively sparing the U fibers and corpus callosum, with extensive coarse calcifications in the basal ganglia, brain stem, and subcortical white matter and the development of parenchymatous cysts.1,2 Ring enhancement of the cyst wall that reflects disruption of the blood- brain barrier was noted both on CT and MR imaging.1 DWI showed increased diffusion in the cysts and the surrounding white matter. This reflects high water content of the abnormal-appearing white matter rather than abnormal myelin. 1H-MR spectroscopy of the cysts showed only lactate peak and no evidence of the normal metabolites of the brain. This suggests energy failure in the parenchyma forming the cyst wall. These imaging findings support increased water content rather than a demyelinating process in the pathophysiology of the disease, which is in accordance with the reports.1,2 The increased water within the white matter—namely, perifocal white matter edema—is likely due to disruption of the blood-brain barrier, which is evident with contrast enhancement in the cyst wall.
During the 4-year-follow-up period, some cysts enlarged gradually and caused compression to the close structures such as ventricules, pons and basal ganglions. Expanding cystic formations may be the consequences of progressive increase in fluid content of the white matter.1,2 This phenomenon was shown in other conditions, such as some intracranial tumors and leukoencephalopathies.4–6 A possible mechanism could be fusion of microcysts containing edema fluid.6 The expanding nature of the cysts in our case may be associated with such a mechanism.
Labrune et al reported the results of histopathologic examination in 2 of 3 patients with LCC1. They noted exuberant proliferation of abnormal small vessels associated with Rosenthal fibers, intense gliosis, and microcalcifications on the specimens. According to Labrune et al, the probable primary pathologic feature is rearrangements involving the microvessels, whereas perivascular foci of calcifications, hyaline deposits, and formation of Rosenthal fibers appeared compatible with secondary changes.1 Nagae-Poetscher et al also presented the histopathogic findings of only one patient.2 On the specimen, despite the presence of macrophages filled with ferric iron deposits around most vessels and few vessels partially calcified, no definite angiomatous changes were revealed. They reported that the absence of the angiomatous changes may have been due to a small tissue sample that included a fragment of the cyst wall and not the surrounding white matter. The histopathologic findings of our patient are compatible with LCC. On the specimen, angiomatous changes and secondary gliosis were the prominent features.
In conclusion, the etiology of the association of the LCC triad remains unknown but probably represents a distinct clinicoradiologic entity. The most striking histopathologic feature is cerebral angiomatous changes. Despite these relatively characteristic imaging and histopathologic findings, no uniformity on clinical features is notable. It may be speculated that older onset age, normal intelligence, and very slow progression, as in our patient, may indicate the presence of the adult form of this rare disease.
References
- Received September 22, 2004.
- Accepted after revision June 6, 2005.
- Copyright © American Society of Neuroradiology









{kind=link}
{kind=link}
{kind=link}
{kind=link}