
Abstract
BACKGROUND: Reversible lesions in the splenium of the corpus callosum (SCC), caused by various agents such as influenza, rotavirus, Escherichia coli, mumps, and adenovirus, were previously defined in a handful of cases. We present 5 cases with transient diffusion restriction of the SCC associated with influenza A virus infection.
MATERIALS AND METHODS: Five patients with influenza-associated encephalitis/encephalopathy and sudden-onset neurologic symptoms following a prodromal flulike episode were examined by MR and diffusion-weighted imaging (DWI).
RESULTS: Three patients, who had drowsiness and new-onset convulsions, recovered spontaneously without any medication. In the other 2 seizure-free patients, 1 had trigeminal neuralgia and headache and the other had facial numbness and left upper monoparesis. All patients had round well-defined ovoid hyperintense splenial lesions (14.94 ± 1.87 mm) on DWI with a significantly low apparent diffusion coefficient (ADC) of 0.41 ± 0.05 × 10−3 mm2/s compared with 0.84 ± 0.01 × 10−3 mm2/s of normal-appearing white matter. In the patient with a motor deficit, additional lesions were found in the cerebral deep white matter. The high signal intensity of the splenial and deep white matter lesions on DWI completely disappeared on follow-up studies, and ADC values also improved, returning to those of normal-appearing white matter on days 8–11. Clinically, all patients completely recovered on days 4–9.
CONCLUSION: A transient lesion of the SCC is a significant but nonspecific finding. It is probably due to edematous and/or inflammatory changes of the SCC. It may be the only detectable change in patients with good prognosis, indicating a clinically mild form of encephalitis/encephalopathy.
Influenza A is the most common upper respiratory tract infectious agent causing flulike symptoms.1 It is especially widespread in winter seasons and can cause epidemics.1–3 Besides Reye syndrome4 and hemorrhagic shock and encephalopathy syndromes,5 it can occasionally cause rapid progressive encephalopathy with high fever, alteration of cognition, and convulsion, which is called influenza-associated encephalitis/encephalopathy (IAEE), including acute necrotizing encephalopathy.1–3,6–8 IAEE is more common and has a poorer prognosis in children than in adults.1,2,9 In clinically mild IAEEs, neurologic symptoms can recover quickly, usually without any specific medication.
The previously described brain lesions in patients with IAEE include restricted diffusion involving the cerebral cortex and subcortical white matter in various localizations; symmetric lesions in the brain stem, basal ganglia, thalamus, and cerebellar white matter with or without brain edema; and mild brain atrophy.3,10–12 Transient restricted diffusion of the splenium of the corpus callosum (SCC) in patients with IAEE was also well defined in previous articles.13,14 However, it is not specific to IAEE and has been reported secondary to various infectious agents, including rotavirus,15 measles,16 herpesvirus 6,17 Salmonella organisms,18 mumps,14 varicella-zoster virus,14 adenovirus,14 O157 Escherichia coli–associated hemolytic-uremic syndrome,19 Legionnaires’ disease,20 and unknown pathogens.14,21 Neither the exact pathophysiology nor the specific site predilection of transient SCC lesions was clear. The most possible causes of these transient lesions of the SCC have been explained as rapidly resolving intramyelinic edema or the influx of inflammatory cells and macromolecules, combined with related cytotoxic edema.13,14
In this study, we describe the imaging findings of 5 patients with IAEE and transient diffusion restriction of the SCC and discuss the possible pathogenesis of these lesions in view of previous reports.
Methods
Patient Population
Five patients, including 3 men and 2 women, ranging from 6 to 41 years of age (mean age, 22.2 ± 12.07 years), with sudden-onset neurologic symptoms following a prodromal flulike episode, underwent contrast-enhanced MR and diffusion-weighted imaging (DWI) to rule out meningoencephalitis. The beginning of the flulike symptoms was taken as day 1, and initiation and resolution of all neurologic symptoms and radiologic findings were expressed in time increments from day 1. Initial MR imaging examinations were performed 2–4 hours after admission to hospital. Patients did not have any medication, especially corticosteroids or antiepileptic drugs, before the MR imaging. The IAEE diagnosis was confirmed by the isolation of influenza A virus from their throat swabs. Follow-up MR imaging in all patients was performed by the same protocol on days 8–11. Informed consent was obtained from all patients for MR imaging studies and for review of patients’ records and images.
All patients were examined by a 1.5T superconducting MR scanner (The New Intera Nova, Philips Medical Systems, Best, the Netherlands) by using a standard quadrature head coil. Axial T1-weighted (TR/TE, 583/15 ms; 1 excitation) spin-echo (SE), T2-weighted (TR/TE, 2295/90 ms; 2 excitations) turbo SE, and fast fluid-attenuated inversion recovery (FLAIR; TR/TE/TI, 8000/100/2000 ms; 1 excitation) images were obtained by using 5-mm section thickness with 1-mm intersection gap and a 256 × 256 matrix size. After intravenous administration of 0.2 mg/kg of gadodiamide (Omniscan), contrast-enhanced T1-weighted SE sequences were also obtained in 3 orthogonal planes.
DWI was performed by using an axial multisection single-shot echo-planar SE sequence (TE, 91 ms; shortest TR ranging from 4200 to 4300 ms; 1 excitation; 1833.3 Hz/pixel bandwidth; echo-planar factor, 89; 22 sections with 5-mm section thickness without intersection gap; matrix size,112 × 256; field of view, 220 × 220 mm in 29.5 seconds). The apparent diffusion coefficient (ADC) maps were calculated on a pixel-by-pixel basis. Standard mean ADC values of each region of interest from lesions and normal-appearing white matter were calculated automatically and expressed in 10−3 mm2/s.
Results
All patients were previously healthy and had no history of seizure, usage of antiepileptic drugs, or any type of vaccination during the last 2 years. Neurologic symptoms became prominent on days 2–5 after the initiation of a prodromal flu episode and high fever (40.3 ± 0.6°C). The new-onset convulsions (on days 2–4) occurred in 3 patients before being admitted to the hospital and resolved spontaneously without any medication. These 3 patients also had drowsiness and some abnormality on electroencephalography. In the other 2 seizure-free patients, 1 had trigeminal neuralgia and headache (on day 2) and the other had facial numbness and left upper monoparesis (on day 5). Rapid-onset neurologic symptoms following a prodromal flu episode were typical for IAEE. In all patients, influenza A virus was isolated from their throat swabs, allowing the diagnosis of IAEE. The influenza genome was not detected in any of their CSF by polymerase chain reaction. Clinically, all patients were completely recovered on days 4–9. The results of blood count, routine biochemistry, and CSF analysis of all patients were in normal limits during illness.
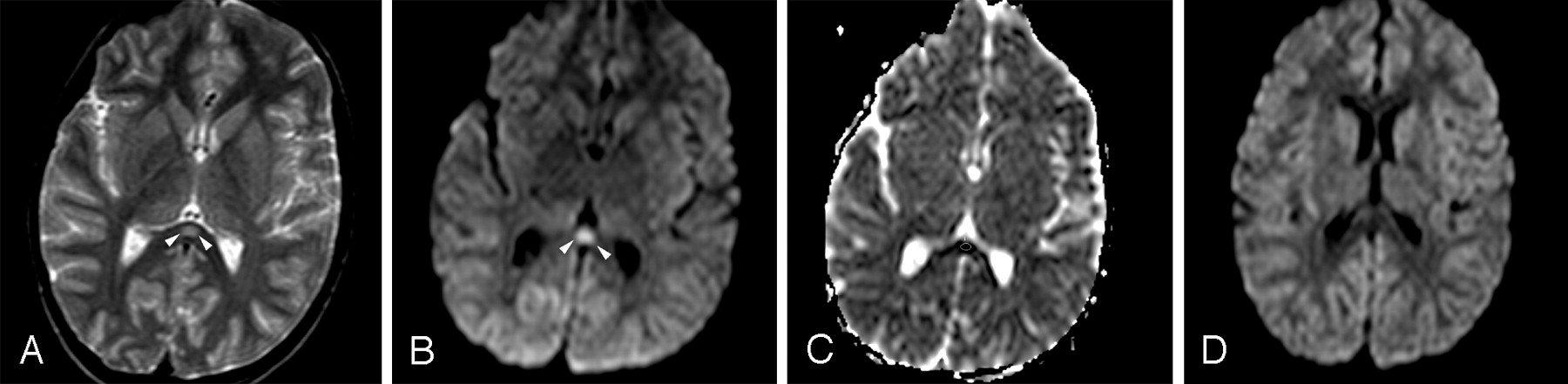
Initial MR imaging examination of each patient was performed on the day that his or her neurologic symptoms developed (on days 2–5). All patients had significant transient lesions in the SCC on their initial MR images (Figs 1A, -C, -E and 2A–C). Lesions were well defined, ovoid, and centrally located in the SCC. The mean diameter of splenial lesions was 14.94 ± 1.87 mm. They were slightly hyperintense on FLAIR and T2-weighted images (Figs 1A, -B and 2A) but not detectable on T1-weighted images. No lesions were enhanced on postcontrast images. All lesions were prominently more hyperintense on isotropic DWI than on T2-weighted and FLAIR images (Figs 1C, -D and 2B). They had significantly lower signal intensity and ADC values (0.41 ± 0.05 × 10−3 mm2/s) than those of normal-appearing white matter (0.84 ± 0.01 × 10−3 mm2/s) (Figs 1E, -F and 2C) on ADC map images. In 1 patient who had mild motor deficits, there were additional lesions in the cerebral deep white matter (Fig 1B,-D, -F). No other signal intensity or diffusion change was detected in the other patients.

Patient 1 (21 years old) with IAEE and sudden onset of facial numbness and left upper monoparesis on day 5. A, Axial T2-weighted image shows a hyperintense well-defined and circumscribed splenial lesion (arrowheads). B, Axial T2-weighted image through the supraventricular region shows patchy high signal intensity of deep white matter lesions (arrows). C and D, Splenial (arrowheads) and deep white matter (arrows) lesions have higher signal intensity on isotropic DWI than those on T2-weighted images. E and F, ADC map images reveal significant restricted diffusion with reduced ADC values obtained from 3 regions of interest of splenial and white matter lesions (0.42 ± 10−3 mm2/s, 0.47 ± 10−3 mm2/s, and 0.51 ± 10−3 mm2/s). G and H, Follow-up isotropic DWI on day 11 shows the reversal of diffusion restriction in both splenial and white matter lesions.
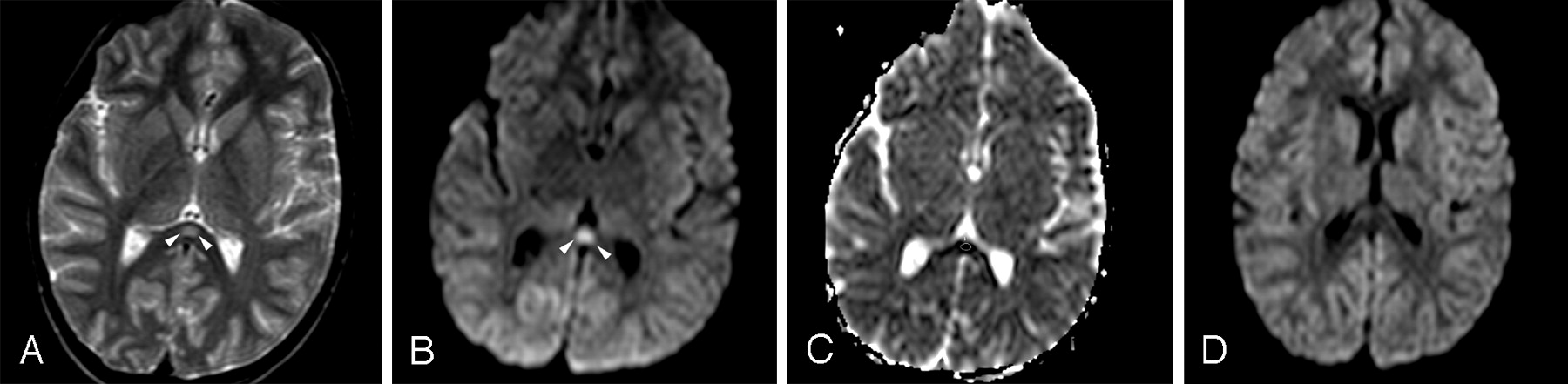
Patient 4 (6 years old) with IAEE and fever and sudden-onset convulsion on day 3. A, Single ovoid well-defined splenial lesion (arrowheads) is slightly hyperintense on the T2-weighted axial image. B, The lesion (arrowheads) has prominently high signal intensity on isotropic DWI. C, ADC value of 0.34 ± 10−3 mm2/s obtained from the region of interest located in the lesion reveals restricted diffusion on ADC map image. D, Follow-up study on day 9 shows complete resolution of diffusion restriction on isotropic DWI.
Follow-up examinations were performed on days 8–11. The high signal intensity of all lesions on DWI disappeared (Figs 1G, -H and 2D), and their ADC values (0.81 ± 0.04 × 10−3 mm2/s) were recovered to normal-appearing white matter (0.84 ± 0.01 × 10−3 mm2/s).
Discussion
IAEE is a complex clinical syndrome, including both encephalitis and encephalopathy.1,6 When there is no evidence of inflammatory change, the term “encephalopathy” is used instead.14 There is probably a continuum and/or an overlap between these clinical syndromes,1,2 so they are generally named together. The onset of neurologic symptoms is usually within a few days to a week after the first signs of influenza A infection and complete recovery occurs within a month as in our patients.1–3,9,14 The clinical findings of our patients were similar to results of previous reports,1,3,9,13,14,22 except those of trigeminal neuralgia. This might be the consequence of a viral route of entry to the brain, because the trigeminal nerve is one of the most promising neuronal pathways of virus within the olfactory nerve.23,24Motor deficits, seen in our 1 patient, were probably due to deep white matter lesions rather than to the splenial lesion itself.
The CSF analyses results of all our patients were within normal limits and free of virus antigens by polymerase chain reaction. The CSF analyses of patients with IAEE often reveal lack of pleocytosis or merely a discrete elevation of mononuclear leukocytes. Protein and glucose contents are usually normal,25 though a slightly increased protein level may be present.1 Positive CSF virus isolation is very rare,10,26,27 either owing to the disappearance of the virus by the time of sampling or to undetectably low amounts of virus in the CSF.1,25
The pathogenesis of IAEE is not clear because of a lack of pathologic correlation in mild forms of the disease. The autopsy findings are mainly from the results of necrotizing encephalopathy (ie, the clinically worst form of IAEE). Autopsy results show that the integrity of the blood-brain barrier (BBB) seems to be an important prognostic factor,10 and the disruption of the BBB promotes neuronal degeneration with severe clinical findings. A toxin-mediated aberrant immune activation23 causing endothelial injury, microvascular angiopathy and perivascular edema,19,20,28,29 or inflammatory cytokine release from virus-stimulated glial cells causing rapid breakdown of the BBB23,25,29,30 can be responsible for the neurotoxic effect. However, direct viral invasion of neurons can also occur, because a positive influenza antigen has been reported in brain tissue.1,25 The clinical severity of IAEE can be moderated by the degree of immune competence of the host, virulence of the agent, route of entry, and coexistence of other predisposing factors such as age, previous vaccination, hypoglycemia, electrolyte imbalance, vitamin deficiency, or seizures.1,17,23,30,31
In the mild form of IAEE, the reversal of restricted diffusion and lack of any significant enhancement without fulminant brain edema suggest a limited direct invasion of neurons, which is not sufficient to cause an immunologic response with resultant rapid breakdown of the BBB. The cytotoxic edema seen in acute cellular energy failure, such as acute arterial infarction, can possibly be the cause of decreased ADC values,12,15 because cytotoxic edema is hardly ever reversible. Intramyelinic edema due to separation of myelin layers seems to be the main contributor of these transient changes.13,14,32 Oster et al33 suggested that reversible restricted diffusion in the SCC is due to transient disruption of energy metabolism and ionic transport, causing reversible myelin vacuolization or intramyelinic edema. Furthermore, autopsy studies of patients with serious neurologic complications have shown that the acute reactive changes, such as congestion and hyperemia without inflammatory infiltration,25,28,34 are more frequent than demyelination34,35 and neuronal degeneration.36
As stated by Tada et al,14 another possible mechanism of transient ADC reduction of SCC is the influx of inflammatory cells and macromolecules, combined with related cytotoxic edema, similar to the changes occurring in multiple sclerosis plaques.37 The transient nature of the lesions suggests that the effect of virus on brain, either inflammatory or edematous, is reversible and may be the only detectable change in patients with good prognosis, a sign of clinically mild encephalitis/encephalopathy.
Differential diagnosis of lesions involving the SCC includes ischemia, infections, posterior reversible encephalopathy syndrome, diffuse axonal injury, multiple sclerosis, hydrocephalus, Marchiafava-Bignami disease, adrenoleukodystrophy, AIDS dementia complex, lymphoma, epilepsy, and antiepileptic drug usage.13,31,33,38–42 The transient feature of the lesion and other clinical and laboratory findings allow one to differentiate the infectious causes from others, but it is not easy to presume the exact infectious agent by clinical and radiologic findings. In latter situation, various infectious agents including influenza,13 rotavirus,15 measles,16 herpesvirus 6,17 Salmonella organisms,18 mumps,14 varicella-zoster virus,14 adenovirus,14 E coli,19 and Legionnaires’ disease20 should be considered in the differential diagnosis.
Another challenging issue is the increased vulnerability of the SCC. Anatomic studies dealing with the corpus callosum demonstrate neither different fiber attenuation nor principally fiber composition in the SCC, compared with other regions of the corpus callosum.43,44 Although the SCC has an arterial supply from the vertebrobasilar system, contrary to other parts of the corpus callosum supplied by the carotid system,45 the absence of any signal-intensity change in the same vascular territory eliminates the vascular etiology.13,14 The special affinity of the receptors on splenial axons or surrounding myelin sheaths to viral antigens or receptors on the antibodies induced by the antigens, expressed by elevated cytokines, can be the cause of this vulnerability, but this theory of Tada et al14 has lack of pathologic correlation. The exact reason for increased predilection of the SCC is still unclear and needs further animal and human research, but it probably remains in debate because of lack of pathologic confirmation of such transient lesions in humans.
The major drawbacks of this study are the lack of histopathologic correlation and low sampling numbers. Rapid resolution of clinical and radiologic findings prevents the biopsy requirement. The rare occurrence of IAEE is the natural cause of the low sampling number.
Conclusion
Although transient ADC reduction of SCC is not pathognomonic for IAEE, it is usually seen in patients with good prognosis, indicating a clinically mild form of encephalitis/encephalopathy. It is more likely due to intramyelinic edema or an inflammatory infiltrate of the SCC rather than a breakdown of the BBB or demyelination. Lack of pathologic correlation in such transient lesions does not allow us to identify the exact nature and pathogenesis of these lesions. Increasing numbers of such cases in the literature allow us to achieve a more reliable conclusion.
References
- Received December 15, 2005.
- Accepted after revision January 25, 2006.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Clinical Features of Cytotoxic Lesions of the Corpus Callosum Associated with Aneurysmal Subarachnoid Hemorrhage
- Brain MR Imaging Findings in Woodhouse-Sakati Syndrome
- Brain Imaging in Cases with Positive Serology for Dengue with Neurologic Symptoms: A Clinicoradiologic Correlation
- Mild encephalopathy with reversible splenial lesion in a patient with influenza A infection--first report in an adult patient in the USA
- Reversible Pancallosal Signal Changes in Febrile Encephalopathy: Report of 2 Cases
- MR Imaging in Novel Influenza A(H1N1)-Associated Meningoencephalitis