
Abstract
The basal ganglia and thalami are paired deep grey matter structures with extensive metabolic activity that renders them susceptible to injury by various diseases. Most pathological processes lead to bilateral lesions, which may be symmetric or asymmetric, frequently showing characteristic patterns on imaging studies. In this comprehensive pictorial review, the most common and/or typical genetic, acquired metabolic/toxic, infectious, inflammatory, vascular and neoplastic pathologies affecting the central grey matter are subdivided according to the preferential location of the lesions: in the basal ganglia, in the thalami or both. The characteristic imaging findings are described with emphasis on the differential diagnosis and clinical context.
Similar content being viewed by others


Introduction
The basal ganglia and thalami are paired grey matter structures, embedded deep in the brain hemispheres and are often referred to as the “central grey matter”. They are involved in the regulation of autonomic, motor, sensory, limbic and endocrine functions, and consequently, their metabolic demand exceeds that of the cerebral cortex in the resting state. Basal ganglia pathologies typically cause movement disorders, but their role extends beyond the extrapyramidal system to memory, emotion and other cognitive functions. The thalamus is a complex hub receiving subcortical sensory and motor input that projects to both the cortex and striatum. Thalamic lesions can cause chronic pain, sensory loss, amnesia, dystonia and other disorders [1].
The terminology can be confusing because of the variety of names and definitions, complex interconnections and interactions. The components of the basal ganglia and the method of naming depend on which aspect (biochemical, embryological or functional) of these complex structures is considered. For the purpose of this review, the term “basal ganglia” is used in the strictest sense (the telencephalic basal ganglia) and refers to the corpus striatum (comprising the caudate nucleus, putamen and globus pallidus). The substantia nigra and subthalamic nucleus originate from the mesencephalon and diencephalon, respectively, and are not considered a part of the basal ganglia in this review. Embryologically and functionally, the caudate nucleus and putamen form a unit, the striatum [2]. The lentiform nucleus is a descriptive collective term for both the putamen and globus pallidus, which are divided by a thin layer of white matter, the lateral medullary lamina.
The thalami, situated along the borders of the third ventricle, originate from the embryonic diencephalon and consist of grey matter nuclei but also of white matter tracts. The difference in embryological origin is also reflected in the arterial blood supply: the basal ganglia are supplied by perforating branches of the anterior cerebral artery, including Heubner’s artery, middle cerebral artery and the anterior choroidal artery, all originating from the internal carotid artery; perforating branches of the posterior cerebral artery and posterior communicating artery supply the thalami. The thalamostriate veins collect the venous drainage of both the basal ganglia and thalami, the blood then flows through the internal cerebral veins and basal veins of Rosenthal, which form the great vein of Galen, and pass into the straight sinus [3].
Magnetic resonance imaging (MRI) is the modality of choice for assessment of pathology involving the deep grey matter nuclei, due to its superior contrast resolution compared to computed tomography (CT). Nonetheless, CT has a distinct role as the reference standard for detection of calcification, while susceptibility-weighted magnetic resonance imaging (SW-MRI) has slightly lower accuracy [4]. However, SW-MRI, and especially the phase information of this sequence, is a valid alternative for CT in the detection of calcifications, when CT is not available or only one examination is possible.
On CT, the basal ganglia and thalamus are normally isodense to the cortex. On T2-weighted and FLAIR images, the thalamus, putamen and caudate nucleus have intensities similar to the cortex, whereas the globus pallidus appears slightly hypointense, due to iron depositions. The higher T1 signal intensity of globus pallidus is mostly due to the higher myelin content but is also caused by calcium deposition. T1 signal intensity increases with concentrations of calcium of up to 30%, but then decreases with higher concentrations [5]. The internal medullary lamina, which divides the globus pallidus in internal and external parts, is not well visualised on currently used clinical MRI scanners (with field strengths 1.5–3 T). The basal ganglia and thalami do not enhance after contrast administration and do not show alteration of diffusivity on apparent diffusion coefficient (ADC) maps. On SW-MRI, the globus pallidus shows a more pronounced hypointensity due to calcium and iron depositions. Increasing mineralisation with age and volume loss occur in all deep grey matter structures [6].
Overview of pathology
It would be an impossible task to include all pathological processes that can affect the central grey matter in this pictorial review. As the goal was to provide an instructive overview of the characteristic imaging patterns that we believe are useful in daily clinical practice, disorders with atypical appearances and those that are exceedingly rare have been excluded, along with some of the commonly encountered entities, primarily lacunar infarcts and metastatic neoplasms. Based on the predominant location of lesions on imaging studies (basal ganglia, thalamus or both), the diseases have been classified into three groups, which were further subdivided according to the type of pathology (primarily inherited metabolic/genetic, acquired metabolic/toxic, inflammatory and infectious, vascular and ischaemic, neoplastic). A summary figure at the end of the paper consists of multiple cropped axial images showing the most characteristic disease patterns involving the basal ganglia and thalami. While this format is simplified and far from perfect, in our opinion, it does allow for a systematic overview of the pertinent imaging findings.
Pathology predominantly affecting the basal ganglia
Inherited metabolic/genetic
Subacute necrotising encephalopathy (Leigh syndrome)
Subacute necrotising encephalopathy (Leigh syndrome) is a progressive mitochondrial neurodegenerative disorder with a severe prognosis that presents in infancy or early childhood. This most common mitochondrial disease is genetically heterogeneous with mutations identified in over 75 genes. Affected infants present with feeding difficulties, psychomotor retardation and ataxia, most commonly during or after a period of metabolic stress. Elevated lactate levels (which may also be found on brain MR spectroscopy) in the arterial blood and/or CSF is a clue to the diagnosis.
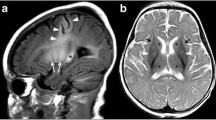
MRI shows bilateral symmetric areas of hyperintense T2 signal in the putamen (which are characteristic but not always present) and/or brainstem (midbrain and medulla, including periaqueductal grey matter) (Figs. 1, 2 and 59) with decreased diffusivity in acute phases due to cytotoxic oedema. The subthalamic nucleus, substantia nigra, caudate nucleus, globus pallidus, dorsomedial thalamus and cerebellar dentate nuclei may also be involved [7, 8]. These MRI signal alterations reflect the spongiform changes and vacuolation in the affected brain structures.

Leigh syndrome. A 6-year-old girl with Leigh syndrome in the acute phase. a Axial T2-weighted image at the basal ganglia level shows bilateral symmetric hyperintensity of the striatum and focal medial thalamic lesions (arrows). There is slightly increased signal intensity of globi pallidi. b Axial T2-weighted image through the midbrain demonstrates hyperintensity of the tectum, periaqueductal grey matter (arrowhead) and bilateral substantia nigra. On MR spectroscopy with echo time of 144 ms, lactate was detected as an inverted double peak on 1.33 ppm (not shown)

Leigh syndrome. A 4-year-old boy with Leigh syndrome. Axial FLAIR image reveals bilateral symmetric hyperintensity in the dorsal putamen. There are additional symmetric brainstem lesions (not shown)
Similar findings can be seen in other mitochondrial disorders, Wilson disease, hypoxic–ischaemic encephalopathy and toxic causes. However, bilateral symmetric T2 hyperintensity involving multiple brainstem structures along with basal ganglia in a child with neurological problems should prompt consideration of Leigh syndrome.
Glutaric aciduria type 1
Glutaric aciduria type 1 is an autosomal recessive metabolic disorder, usually presenting in infants. The disease frequently starts abruptly with focal or generalised seizures following an infection or immunisation. Symptoms then evolve to psychomotor regression and dystonia or choreoathetoid movements. Macrocephaly is usually present at birth.
MRI shows a characteristic frontotemporal atrophy with enlarged Sylvian fissures (“bat wing” dilatation), subdural hygromas and delayed myelination (Figs. 3, 4 and 59). There is increased T2 signal in the caudate nucleus and the putamen, with swelling and reduced diffusivity in the acute phases, evolving to atrophy over time [9, 10]. Confluent white matter signal alterations and subependymal nodules along the lateral ventricles (Fig. 3b) are present in some cases. Due to the presence of subdural hygromas, this condition may be a mimicker of non-accidental traumatic injury. However, the characteristic associated abnormalities should alert the radiologist to the possibility of a metabolic disease [11].

Glutaric aciduria type 1. A 14-year-old male with glutaric aciduria type 1. a On axial T2-weighted image, there is bilateral symmetric hyperintensity of the posterior lentiform nuclei (arrows). b Confluent fronto-temporo-parietal areas of high signal intensity (white arrowheads) are better seen on the coronal T2-weighted image, which also demonstrates subependymal nodules (black arrowheads) along the superior aspect of the frontal horns

Glutaric aciduria type 1. A 20-month-old boy with glutaric aciduria type 1. a Axial T2-weighted image shows subcortical areas of delayed myelination, primarily frontal (arrowheads), along with more prominent bilateral symmetric hyperintensity of the globi pallidi and posterior putamina (arrows). b ADC map reveals lower signal intensity of the lentiform nuclei (arrows), consistent with decreased diffusivity. There are also bilateral wide opercula/enlarged Sylvian fissures (*), best seen on the left side in a
Wilson disease
Wilson disease (WD) or hepatolenticular degeneration is an autosomal recessive condition caused by ATP7B gene mutations that result in errors of copper transport and accumulation in the liver and deposition in other sites, including the brain. The diagnosis is typically established by the presence of low ceruloplasmin levels and Kayser–Fleischer ring around the iris, supported by high urine copper and confirmed by genetic testing. Extrapyramidal and behavioural symptoms typically begin in the second or third decade of life; however, it may manifest in older people and in young children [12,13,14]. On T2-weighted and FLAIR images, there is bilateral symmetric deep grey matter signal alteration, commonly a concentric-laminar hyperintensity. High T2 signal in the putamen (particularly along the outer rim) is most frequently observed (Figs. 5a and 59), followed by changes of the caudate nucleus, globus pallidus, thalamus (sparing the medial portion) and brainstem. Reduced diffusivity may sometimes be present in very early phases; however, increased diffusion is more typical. Symmetric T1 hyperintensity of globus pallidus is common (Figs. 5b and 59), likely resulting from liver failure. Hyperintensity of the midbrain with sparing of red nuclei, portion of substantia nigra and superior colliculi produces the “face of the giant panda” sign, which is virtually pathognomonic for WD but present in a minority of patients.

Wilson disease. A 6-year-old girl with headache, tremor and liver failure. a Axial T2-weighted image reveals bilateral symmetric hyperintensity of the putamen and caudate nucleus, more prominent peripherally. There is a subtle increase in signal intensity in bilateral globus pallidus and lateral thalamus. b Axial T1-weighted image at a slightly lower level shows bilateral mild decrease in signal intensity of the putamen and hyperintensity of globus pallidus (arrows)
The signal alterations regress with response to copper-chelating treatment [12,13,14]. Similar neuroimaging findings may be seen in Leigh disease, methanol toxicity and metabolic acidosis. However, bilateral basal ganglia with or without thalamic or brainstem lesions on MRI are highly suggestive of WD in the appropriate clinical setting.
Huntington disease
Huntington disease (HD) is an autosomal dominant neurodegenerative disease caused by a mutation (an elongation of CAG repeats, coding for glutamine) in the gene which encodes huntingtin protein. The mutant huntingtin protein aggregates in axonal terminals and causes neuronal cell death, which is most pronounced in the spiny GABA-ergic neurons of the striatum. The disease is characterised by a triad of motor, cognitive and psychiatric symptoms, showing an unrelenting progressive course over 15–20 years. An average age of onset is 40 years, though cases of HD have been diagnosed in patients ranging from 2 to 87 years. Adult-onset HD typically presents as a hyperkinetic, whereas the juvenile form as a hypokinetic motor disorder [15, 16].
The characteristic imaging finding of HD is bilateral striatal (caudate nucleus and putamen) atrophy, particularly involving the heads of the caudate nuclei. This leads to symmetric enlargement of the adjacent frontal horns with flattening of their lateral contour, giving the “boxed-out” appearance (Figs. 6 and 59). The intercaudate distance exceeds 20 mm, whereas normal values range from 10 to 14 mm. A mild associated T2 hypointensity due to iron deposition may be noted in the adult form of the disease. In the juvenile-onset HD, on the other hand, high T2 signal is frequently observed in the caudate nucleus and putamen (Fig. 7). Diffuse cerebral atrophy with white matter volume loss, generally more pronounced in the frontal lobes, is observed in advanced stages of the disease. Even years before symptoms become overt, mutation carriers show subtle but progressive striatal and cerebral white matter atrophy, demonstrated by volumetric MRI [15, 16].

Huntington disease. A 36-year-old male patient with a family history of Huntington disease and choreoathetosis. There is severe atrophy of the caudate nucleus head on coronal post-contrast T1-weighted image with frontal horns of the lateral ventricles demonstrating “boxed-out” appearance (arrowheads). There is also atrophy of the frontal lobes

Huntington disease. A 17-year-old patient with a juvenile form of the disease. Axial T2-weighted image shows a reduced size of bilateral caudate head (arrowheads) and putamen (arrows). The associated hyperintensity is more prominent in the putamina
Neurodegeneration with brain iron accumulation
Neurodegeneration with brain iron accumulation (NBIA) is a group of inherited neurologic disorders characterised by abnormal accumulation of iron in the basal ganglia. The disease onset ranges from infancy to adulthood, while progression can be rapid or slow with long periods of stability. These patients present with dystonia, spasticity, parkinsonism and neuropsychiatric disturbances, along with MRI evidence of iron accumulation. Generalised cerebral and cerebellar atrophy are frequently also present [17, 18].
The pattern of iron distribution varies among these disorders, with additional features helping in the differential diagnosis. The “eye of the tiger” sign, referring to hypointense globus pallidus with a central hyperintensity on T2-weighted images (Figs. 8 and 59), is almost pathognomonic for pantothenate kinase-associated neurodegeneration (PKAN, formerly called Hallervorden–Spatz disease), the most common disorder (up to 50% of all NBIA cases). Furthermore, in mitochondrial membrane protein-associated neurodegeneration (MPAN), T2 hyperintense streaking of the medial medullary lamina between the internal and external globus pallidus is present in some patients. Finally, cystic changes and cavitations in caudate and putamen are unique to neuroferritinopathy [17, 18].

PKAN. An 8-year-old boy with pantothenate kinase-associated neurodegeneration. a Bilateral symmetric hyperintense foci in the anteromedial globus pallidus are surrounded by lower signal intensity on FLAIR image (arrows), giving the typical “eye of the tiger” sign. b There is a corresponding mild hyperintense signal on T1-weighted image
Note that “eye of the tiger” sign may also be observed in Wilson disease, atypical parkinsonism or organophosphate poisoning and that it may even be a normal finding on 3 T scanners. On the other hand, the MRI imaging features may precede symptoms in patients with NBIA disorders [17].
Acquired metabolic/toxic
Carbon monoxide poisoning
Exposure to carbon monoxide (CO) is the most common cause of human poisoning around the world, caused by incomplete combustion of carbon-based fuels. Following inhalation, CO binds to haemoglobin with a 250-fold stronger affinity than oxygen, thereby reducing oxygen carrying capacity and leading to subsequent tissue hypoxia. Furthermore, CO prevents cellular uptake of oxygen, thus impairing the mitochondrial respiratory function, and promotes the formation of oxygen free radicals. Neurological symptoms of CO poisoning can manifest not only immediately but also as late as 2 to 6 weeks after successful initial resuscitation as delayed neurological sequelae.
Bilateral focal globus pallidus lesions that are hypodense on CT and hyperintense on T2-weighted, FLAIR and DWI (Figs. 9, 10 and 59) are the very characteristic imaging findings, with the corresponding reduced diffusivity seen in the acute phase. In some patients, there is a central or peripheral area of low signal on gradient echo (GRE) T2*-weighted images/SW-MRI, occasionally accompanied with corresponding T1 hyperintensity, which is indicative of haemorrhage. In the acute phase, a patchy and/or peripheral contrast enhancement may also be seen. A very common imaging finding in the early stages is that of bilateral cerebral white matter T2 and DWI hyperintensities [19]. Involvement of the putamen, caudate nucleus, thalamus, hippocampus, cerebellum and substantia nigra may occasionally be present, typically with more subtle lesions. On the other hand, isolated abnormalities of bilateral globus pallidus lesions may occur in patients with methanol intoxication and hypoxic–ischaemic encephalopathy (HIE). Interestingly, it seems that the cerebral white matter signal alterations may actually represent the most common MRI finding of CO poisoning, instead of the characteristic lesions of the globi pallidi. The white matter lesions are also considered primarily responsible for the chronic symptoms [20].

Carbon monoxide (CO) poisoning. A 37-year-old fire victim with headache, nausea and vomiting. Axial CT shows bilateral focal hypodense lesions within globus pallidus (arrows)

CO poisoning. Acute CO intoxication from malfunctioning heater. Axial FLAIR image shows hyperintense oval lesions in bilateral globus pallidus with central hypointensity (suggestive of small haemorrhage). There are also white matter hyperintensities (arrowheads), more extensive in the centrum semiovale (not shown)
Bilirubin encephalopathy (kernicterus)
Kernicterus is the chronic form of an acquired bilirubin encephalopathy that develops over months to years in infants who survive the acute stages of bilirubin encephalopathy in the neonatal period. The neuroimaging hallmark in the chronic phase is the presence of bilateral symmetric high T2 signal in the globus pallidus (Figs. 11 and 59) and subthalamic nucleus [21]. Increased T2 signal may also be seen in the substantia nigra or in the cerebellar dentate nucleus, while T1 signal is variable. On the other hand, MRI in the acute to subacute phase characteristically demonstrates increased T1 signal of globi pallidi and subthalamic nuclei. In contrast to hypoxic–ischaemic encephalopathy, there is no T2 hyperintensity or diffusion abnormality and the thalami and putamen are of normal appearances. In term neonates, a key differential is normal myelination since the globus pallidus and subthalamic nucleus are spontaneously hyperintense on T1-weighted sequences. Of note, 3D GRE T1-weighted images at 3 T demonstrate a particular propensity for hyperintense signal in these regions with normal myelination [21].

Kernicterus. A 4-year-old boy with chronic bilirubin encephalopathy. T2-weighted image reveals symmetric hyperintensity of the globi pallidi. Symmetric increased signal was also present at the dentate and subthalamic nuclei (not shown)
Chronic hepatic encephalopathy/manganese toxicity
Chronic hepatic encephalopathy (CHE) most commonly develops in the setting of cirrhosis and portal hypertension or portosystemic shunts. It includes a spectrum of neuropsychiatric abnormalities caused by liver dysfunction.
Bilateral symmetric globus pallidus hyperintensity on T1-weighted images is the characteristic finding in patients with CHE (Figs. 12 and 59), without corresponding abnormality on CT or T2-weighted MRI. High T1 signal may also be seen in the bilateral cerebellar dentate nucleus, substantia nigra, subthalamic nucleus and the tectum. The accumulation of manganese (Mn) is considered responsible for the T1 signal alteration. The serum concentration of Mn in cirrhotic patients is three times higher than in healthy individuals, due to inadequate liver elimination. Diffuse white matter T2 hyperintensity with preferential involvement of the corticospinal tracts and subcortical focal bright T2 lesions may also be present [22].

Chronic liver failure/Mn toxicity. A 49-year-old female with known hepatic cirrhosis presenting with new onset tremor. On axial T1-weighted image, there is symmetric hyperintensity of the globi pallidi. The increased signal is extending to the tegmentum of the midbrain and dentate nuclei (not shown). There is also increased T2 signal intensity in the internal capsules (not shown)
Restoration of liver function, such as with liver transplantation, leads to improvement of the MRI abnormalities, including T1 hyperintensities, increased T2 signal of the white matter, as well as spectroscopy findings (decreased myo-inositol levels and elevated glutamine/glutamate complex) and return to normal appearances [22]. Note that findings of Mn deposition can be seen in patients with longstanding total parenteral nutrition, Crohn’s disease and long-term use of proton pump inhibitors in the treatment of gastric ulcers.
Gadolinium deposition
The symmetric T1 hyperintensity of the globus pallidus, along with cerebellar dentate nucleus, is also commonly seen with deposition of gadolinium (Gd) following multiple contrast-enhanced MRI scans, primarily with linear gadolinium-based contrast agents (GBCA) [23, 24]. This phenomenon was first reported in 2014 and there are currently no known neurologic, neuropsychological or other clinical consequences resulting from Gd deposition. The MRI appearances may not be distinguishable from those with chronic hepatic encephalopathy or other causes of Mn accumulation.
Methanol poisoning
Methanol intoxication usually occurs after oral ingestion of industrial liquids or consumption of contaminated alcoholic beverages, often occurring in mass poisonings. The most striking acute symptoms after ingestion are visual disturbances. Other symptoms are delayed up to 24 h, as methanol is metabolised by alcohol dehydrogenase to formic acid in the liver, causing severe metabolic acidosis. The mainstay of treatment is intravenous administration of ethanol, due to its higher affinity for alcohol dehydrogenase. Methanol intoxication has high mortality and leads to long-term visual sequelae and severe brain damage in survivors [25, 26].
Characteristic haemorrhagic or non-haemorrhagic bilateral symmetric lesions of the putamen are seen 24 to 36 h after methanol ingestion. The putamina are hypodense on CT and hyperintense on T2-weighted MR images (Figs. 13 and 59), commonly with T1 hyperintensity reflecting haemorrhage. Reduced diffusivity is found in the acute phase and there may be (usually peripheral) contrast enhancement. The caudate nuclei and globi pallidi may also be involved, as well as the cortex [25, 26]. Bilateral putaminal haemorrhage, especially with associated frontal and insular white matter lesions (Figs. 13 and 59), is almost pathognomonic for acute methanol poisoning, while isolated symmetric involvement of putamen (and caudate nucleus) may be seen in patients with uremic encephalopathy and other metabolic acidoses.

Methanol poisoning. A 31-year-old man a few days after ingestion of home-made liquor. a Axial FLAIR image shows bilateral symmetric hyperintensity with mild swelling of the putamen and frontal white matter. The signal alteration is accentuated along the lateral putaminal margin (arrowheads), and there are a few additional smaller hyperintense areas. b ADC map at a slightly lower level reveals high diffusivity of the putamina (arrowheads) and reduced diffusion of the affected white matter (arrows), predominantly frontal. Images courtesy of Claudia Godi
Uremic encephalopathy and metabolic acidosis
Uremic encephalopathy is a rare complication of end-stage renal failure with intracranial accumulation of uremic toxins.
In these patients, MRI frequently shows bilateral symmetric swelling of the putamina and globi pallidi with hyperintensity on T2-weighted and FLAIR images. Additionally, there is a rim of even brighter signal intensity that extends from the posterior aspect of the putamen laterally along the external capsule, while its medial arm spreads along the internal capsule and then bifurcates anteriorly into two slightly less hyperintense branches, corresponding to the external and internal medullary laminae (Fig. 14a and 59). The lentiform nucleus is thus delineated and divided into three sections—putamen, globus pallidus interna and globus pallidus externa, resembling a fork. This characteristic imaging appearance has been hence named “lentiform fork” sign [27]. ADC maps demonstrate peripheral bright signal of increased diffusivity along the “fork” with variable appearances centrally, frequently containing dark areas of reduced diffusivity (Fig. 14b). There is no associated contrast enhancement and the caudate nucleus may also be involved. Symptoms and imaging findings improve with renal transplantation or renal dialysis. The lentiform fork sign may be found in other conditions with metabolic acidosis and in patients with diabetes without acidosis such as with metformin-associated encephalopathy [28].

Uremic encephalopathy/metabolic acidosis. A 2-year-old girl admitted with vomiting and diarrhoea for 1 week. Laboratory findings confirmed haemolytic uremic syndrome. a Axial FLAIR image shows bilateral symmetric swelling and increased signal of the central and posterior putamina with a more prominent hyperintense rim along the medial and lateral margins (arrows)—the characteristic “lentiform fork” sign of uremic encephalopathy and metabolic acidosis. There is also a small white matter signal alteration. b The lesions show centrally reduced (arrowheads) and peripherally (along the medial and lateral arms of the putamina) increased diffusivity (arrows) on the corresponding ADC map
Hypoglycaemia
A sudden decrease in the serum glucose level (such as in diabetic patients or with exogenous administration of insulin) may cause coma, but most patients regain consciousness after glucose infusion. Hypoglycaemic encephalopathy develops in extended and severe hypoglycaemia and can cause irreversible brain damage, resulting in a persistent vegetative state or death.
Three patterns of abnormalities may be present on MRI: selective white matter involvement, selective grey matter involvement or both. Additionally, the lesions can be diffuse or more focal, bilateral, asymmetric or even unilateral. The abnormalities have been reported in the cerebral cortex, basal ganglia, hippocampi, subcortical white matter, posterior limb of the internal capsule, and splenium of the corpus callosum with sparing of the thalamus, cerebellum and brainstem. The earliest change in severe hypoglycaemia is reduced diffusivity, due to cell swelling and cytotoxic oedema. The extent of the abnormalities depends on severity and duration of hypoglycaemia with diffuse and extensive DWI lesions predicting a poor neurologic outcome. Sparing of the thalamus allows differentiation from hypoxia, in which the thalami are most commonly affected (Figs. 15 and 59) [29].

Hypoglycaemia. A 77-year-old woman with diabetes mellitus following a suicide attempt with insulin overdose, resulting in hypoglycaemic coma. a The entire visualised cerebral cortex and striatum show bilateral hyperintense signal on axial FLAIR image. b Corresponding ADC map reveals reduced diffusivity of the involved grey matter. The globi pallidi and thalami (*) are spared
Transient hypoglycaemia is relatively common in neonates during the peripartum. When abnormalities are present on imaging, there is characteristic bilateral involvement of the white and grey matter in the parietal and occipital lobes [30].
Hyperglycaemia
The pathological process known as non-ketotic hyperglycaemia with hemichorea-hemiballismus, which describes the clinical presentation, is characteristically found in patients with (poorly controlled) diabetes mellitus, more commonly in elderly females of Asian descent. However, the sudden onset of choreiform and ballistic movements of unilateral extremities may occasionally be the initial and only manifestation of type 1 diabetes in younger people, including adolescents [29].
CT demonstrates hyperdensity of the putamen, typically contralateral to the side of the patient’s symptoms. This finding is, in contrast to essentially all other metabolic or toxic disorders, characteristically and in the vast majority of cases unilateral (and usually strikingly asymmetric in rare bilateral cases). There is corresponding abnormal T1 hyperintensity of the putamen on MRI, with or without involvement of the caudate nucleus and globus pallidus (Figs. 16 and 59). The affected areas demonstrate variable appearance on DWI, ADC, GRE T2*-weighted (or SW-MRI) and T2-weighted images. Imaging features are usually reversible, though they lag behind clinical improvement [29, 31, 32].

Hyperglycaemia. A 79-year-old woman presenting with left-sided hemichorea-hemiballismus. Laboratory findings confirmed non-ketotic hyperglycaemia in this patient with known diabetes. a Axial CT image shows high attenuation of the entire right putamen (arrow) and caudate head (arrowhead) without expansion. b MRI demonstrates hyperintensity of the right putamen on T1-weighted imaging
Inflammatory/infectious
Autoimmune encephalitis
Autoimmune encephalitis refers to a group of immune-mediated pathologies directed to several neuronal antigens, with or without an underlying malignancy. In the first case, the antibodies are usually against intracellular antigens, while in the second case, the antibodies are against surface receptors. Regardless of aetiology or antibody profile, there is a clear predilection for antigens in the limbic system. However, entities targeting antigens in the basal ganglia have also been described [33]. Clinical presentation is generally gradual, evolving over a few weeks and psychiatric symptoms are common, along with movement disorders and seizures.
Anti-CV2/CRMP5 (collapsin response mediator protein 5) encephalitis, which is frequently associated with lung cancer and thymoma, typically presents with chorea and behavioural disorders. In some patients, MRI shows a prominent hyperintensity of the striatum (putamen and caudate nucleus) and claustrum on T2-weighted and FLAIR images (Figs. 17 and 59), without diffusion abnormality, which helps in differentiation from Creutzfeldt–Jakob disease. Anti-CV2 encephalitis is a diagnosis of exclusion and the differential diagnosis should include various intoxications and metabolic acidosis [33,34,35].

Autoimmune encephalitis. A 46-year-old man with gait instability, behavioural disorder and chorea. Axial T2-weighted image demonstrates bilaterally symmetric hyperintensity and mild swelling centred at the putamen (arrows) and caudate nucleus (arrowheads). A slight symmetric increase in signal intensity is present at the cingulate gyri and posterior thalami. Anti-CV2/CRMP5 antibodies tested positive and metastatic lung cancer was eventually diagnosed
Cryptococcosis
Manifestations of cryptococcal CNS infection are variable and include gelatinous pseudocysts, granulomas (cryptococcomas) and meningoencephalitis. Imaging studies, especially CT, may be normal or show ventriculomegaly only. The organisms extend from the basal cisterns into the brain via the perivascular spaces, resulting in cyst-like structures filled with the fungi and their gelatinous capsular material. These characteristic gelatinous pseudocysts are well-demarcated lesions located preferentially in the basal ganglia, ranging from a few millimetres to several centimetres in size [36, 37].
The pseudocysts are non-enhancing, CSF-like on CT as well as on T2- and T1-weighted MRI, however hyperintense on FLAIR images (Figs. 18a and 59). The finding of multiple cysts/dilated perivascular spaces in an immunosuppressed patient is highly suspicious for cryptococcal infection. Cryptococcomas, on the other hand, are T2 hyperintense intra-axial nodules usually located in the deep grey matter that show mild contrast enhancement (Fig. 18b) and may have surrounding oedema [36, 37]. While gelatinous pseudocyst and cryptococcomas are the most common findings in HIV-infected patients, radiological meningitis is the predominant MRI abnormality of cryptococcal meningoencephalitis in HIV-negative individuals [38].

Cryptococcosis. HIV-positive patient with headaches, nausea and vomiting. a Axial FLAIR image shows asymmetric patchy hyperintense lesions in bilateral basal ganglia. b There are focal areas of enhancement within these lesions on corresponding post-contrast T1-weighted image
Vascular/ischaemic
HIE frequently involves the basal ganglia as well as the thalami and is therefore discussed in the “Pathology involving both the basal ganglia and thalami” section.
Neoplastic
Germinoma
Intracranial germinomas predominantly arise in midline supratentorial locations, primarily the suprasellar and pineal regions; however, about 6–10% occur in off-midline structures, usually the basal ganglia (sometimes the thalamus and very rarely the posterior fossa structures). Basal ganglia germinomas are notoriously difficult to diagnose because of the non-specific clinical presentation and subtle abnormalities on initial imaging studies. The lesions may be unilateral or bilateral and show only a mild hyperdensity on CT, along with a slight hyperintensity on T1- and T2-weighted MR images, without mass effect or enhancement. If such subtle findings are bilateral, they may be especially hard to notice. SW-MRI (or GRE T2* images) are particularly helpful, showing the neoplasms as much more obvious hypointensities in the globus pallidus and putamen. On the other hand, germinomas may be accompanied with cystic change, calcification or haemorrhage and even a prominent mass effect. Ipsilateral cerebral and/or brain stem hemiatrophy (Figs. 19 and 59), representing Wallerian degeneration, occurs in later stages with unilateral lesions [39, 40].

Germinoma. A 14-year-old boy with cognitive and behavioural changes, headaches and progressively worsening weakness of the right arm and leg. Axial T2-weighted image shows left greater than right hyperintense lesions in the basal ganglia with prominent peripheral hypointensity (arrows). There is also left hemiatrophy with widened Sylvian fissure (*) and atrium of the lateral ventricle (**). Courtesy of Andrea Rossi
Pathology predominantly affecting the thalami
Inherited metabolic/genetic
Lysosomal storage disorders
Gangliosidoses are a group of recessively inherited storage disorders caused by lysosomal enzyme deficiencies. Both GM1 and GM2 (also known as Tay–Sachs and Sandhoff diseases) are classified into three types: infantile, juvenile and adult chronic. The infantile forms are most common and most severe. Patients present with rapidly evolving motor weakness, developmental delay, deafness and blindness in the first 6 months of life, often with death by 1–3 years of age. The juvenile and adult chronic forms have a different clinical presentation with motor disturbances and intellectual disabilities [41]. Neuroimaging in patients with infantile gangliosidoses is similar in GM1 and GM2 subtypes. Symmetrically hyperdense thalami on CT with corresponding increased T1 and low T2 signal (Figs. 20 and 59) are characteristic. Other features include mild T2 hyperintensity of the striatum as well as diffuse hypomyelination with sparing of the corpus callosum. The juvenile and adult forms mostly present with cerebellar atrophy.

Lysosomal storage disorders—GM1 and GM2 gangliosidoses. A 17-month-old girl with the infantile form of gangliosidosis type 1. a There is bilateral hyperintensity of the thalami (arrows) on T1-weighted image. b T2-weighted image shows corresponding low signal intensity of the thalami (arrows). There is also diffuse hypomyelination of the supratentorial white matter (with low T1 and high T2 signal intensity) and plagiocephaly
Other lysosomal storage disorders can also show bilateral thalamic CT hyperdensity and T2 hypointensity on MRI, primarily Krabbe disease and neuronal ceroid lipofuscinosis [42,43,44,45]. On the other hand, “pulvinar sign” representing unilateral or bilateral T1 hyperintensity of the thalamic pulvinar was initially thought to be a common and pathognomonic sign of Fabry disease (FD), but due to its low incidence (in around 3% of patients) and low specificity, it is no longer considered a characteristic imaging finding of FD [46].
Acquired metabolic/toxic
Toluene toxicity/solvent abuse
Toluene (methylbenzene) is generally the main and most toxic ingredient of industrial solvents as well as many household products, such as glue, gasoline and finger nail polish. While the exposure may be occupational (construction work with paint, employees in fingernail salons and production of gasoline), the recreational abuse (“sniffing glue”) has become a worldwide health and social problem. The mechanism of toxicity is not well understood, despite the high prevalence and serious consequences of toluene-induced encephalopathy.
The imaging studies are typically unremarkable in the acute poisoning. The abnormalities on MRI are found after prolonged chronic abuse (for at least a year, usually a few years) and are irreversible at the time of detection. The characteristic findings are low T2 signal of the thalami and other grey matter, poor differentiation of the cortex from the subcortical white matter, ill-defined white matter T2 hyperintensity and global brain atrophy (Figs. 21 and 59). The characteristic bithalamic low T2 signal may be due to demyelination with secondary deposition of iron and/or to direct accumulation of toluene within the lipid membranes. The duration of abuse correlates with the severity of the imaging findings [47].

Toluene toxicity. A 21-year-old male with organic solvent abuse for 5 years. Axial T2-weighted image shows a prominent hypointensity of both thalami (arrows). Also seen is mildly decreased signal intensity of the left globus pallidus, generalised atrophy and diffusely increased signal of the white matter with loss of grey–white matter distinction. Case courtesy of Dr. Chee Kok Yoon, Radiopaedia.org, rID: 17082
Wernicke encephalopathy
Wernicke encephalopathy (WE) is an acute, initially reversible neuropsychiatric disorder caused by vitamin B1 (thiamine) deficiency, typically presenting with ocular abnormalities, ataxia and confusion. It usually occurs in the setting of poor diet, classically with alcoholism, which is the most common cause of WE in adults. However, non-alcoholic WE, which can result from gastrointestinal tract diseases and/or surgery, hyperemesis gravidarum and other conditions, is considered underdiagnosed. While inadequate thiamine intake is the cause in children with prolonged malnutrition, WE is becoming increasingly prevalent in paediatric patients with malignancies and prolonged intensive care unit stays.
MRI shows typical symmetric signal alterations along the medial thalami, periaqueductal grey matter, quadrigeminal plate and mammillary bodies (Figs. 22, 23 and 59). The dorsal medulla, cranial nerves, cerebellum and perirolandic cortex are also affected in some patients, typically with non-alcoholic WE [48]. The lesions are hyperintense on DWI, FLAIR and T2-weighted images, hypointense to isointense on T1-weighted images and showing some degree of contrast enhancement in around a half of the cases. Diffusivity varies from decreased to increased and may be reversible [49]. WE is a readily treatable severe condition, which should be suspected and diagnosed on MRI.

Wernicke encephalopathy (WE). A 38-year-old female with history of gastric bypass, presenting with somnolence and ophthalmoplegia after 3 days of vomiting and Wernicke encephalopathy. a, b Axial FLAIR images demonstrate hyperintensity involving bilateral medial thalami along the third ventricle, periaqueductal grey matter (arrowhead) in the midbrain and the mammillary bodies (arrows)

WE. A 49-year-old male with WE due to chronic alcoholism. a Axial DWI shows the characteristic bilateral symmetric hyperintensity along the dorsomedial thalami (arrows). b There is no diffusion abnormality on the corresponding ADC map
Metronidazole-induced encephalopathy (MIE) is a rare complication of this widely used antimicrobial medication, with clinical presentation that can simulate WE. One of the possible mechanisms of metronidazole toxicity is through production of inhibitory thiamine analogue metabolites. The characteristic MR finding is bilateral T2 hyperintensity of the cerebellar dentate nuclei, along with involvement of splenium of the corpus callosum, periaqueductal grey matter and dorsal brainstem. The signal abnormality of the periaqueductal grey matter and dorsal midbrain may be indistinguishable from WE; however, lesions of the mammillary bodies and medial thalami are extremely rare in MIE, while lesions of the dentate nuclei are quite unusual for WE. Cases of possible combined WE and MIE have also been described [50, 51].
Acute hyperammonaemic encephalopathy
Acute hyperammonaemic encephalopathy is a life-threatening condition that is most commonly caused by severe liver disease, but it may also occur with non-cirrhotic conditions causing increased ammonia production (such as infections by urea-producing bacteria and multiple myeloma) or decreased ammonia elimination—primarily genetic disorders of urea cycle enzymes, portosystemic shunts and drugs (such as paracetamol and valproate). Patients present with progressive drowsiness and seizures, secondary to brain oedema and intracranial hypertension. Acute hyperammonaemic encephalopathy can progress to coma and death or lead to long-term sequelae, including intellectual impairment [50].
The characteristic MRI findings are bilateral symmetric signal abnormalities in the cortex of the insula and cingulate gyrus, usually accompanied by thalamic involvement (Figs. 24, 25 and 59). The affected areas of cortical and deep grey matter show hyperintensity on T2-weighted images, which is more striking on FLAIR and DWI, while decreased diffusivity may be present on ADC maps. The basal ganglia are usually spared, along with perirolandic and occipital cortices [52, 53].

Acute hyperammonaemic encephalopathy. A 26-year-old man was found unconscious, with upper motor neuron signs, clonus and drowsiness on exam. Biochemical exam demonstrated hyperammonaemia due to valproate toxicity. Coronal FLAIR image reveals bilateral symmetric high signal intensity involving the cortex of insula and cingulate (arrows), thalami (arrowheads) and hippocampi. Courtesy of Sumeet Kumar

Acute hyperammonaemic encephalopathy. A 60-year-old alcoholic patient presenting with confusion and drowsiness due to decompensated liver cirrhosis. a There is bilateral symmetric cortical and thalamic (arrows) hyperintensity on DWI. The cortical signal alteration is most prominent in the cingulate gyri and insulae, while the occipital lobes and basal ganglia are spared. The perirolandic cortex is also spared (not shown). b Corresponding T1-weighted image shows bilateral high signal of globus pallidus (arrowheads), indicative of chronic liver failure. Case courtesy of Dr. Andrew Dixon, Radiopaedia.org, rID: 39037
Inflammatory/infectious
Thalamic involvement, along with white matter lesions, is commonly encountered in children with acute disseminated encephalomyelitis (ADEM). Basal ganglia may also be affected and ADEM is therefore discussed in the “Pathology involving both the basal ganglia and thalami” section.
Acute necrotising encephalitis
A subset of encephalitides associated with respiratory viruses present as acute necrotising encephalitis (ANE), a rapidly progressive encephalopathy that is typically involving bilateral thalami. In contrast to ADEM, the inflammation is characteristically absent, whereas necrosis and haemorrhage are the predominant findings. ANE is usually preceded by a virus-associated febrile illness, most commonly influenza but also other agents including coronavirus disease 2019 (COVID-19), followed by rapid deterioration [54,55,56]. This entity occurs predominantly, but not exclusively in young children and has also been known as acute necrotising encephalitis in children (ANEC). ANE may occur sporadically; however, there are recurrent and familial forms associated with mutations in RANBP2 gene [54].
The very characteristic imaging findings are bilateral symmetric concentric haemorrhagic lesions of the thalami, best seen on SW-MRI/T2* images, with surrounding oedema, usually accompanied by centrally low and periferally increased diffusivity (Figs. 26 and 59). This imaging appearance may also be seen with deep venous thrombosis; however, the internal cerebral veins are patent in patients with ANE. The brainstem and subinsular areas, as well as the cerebellum and basal ganglia, can also be affected [54, 55].

Acute necrotising encephalitis (ANE). A 2-year-old girl with drowsiness following respiratory viral infection. a Axial ADC map shows swelling of both thalami with heterogeneous diffusivity—primarily central low and peripheral high signal. b The central areas show signal loss (arrows) on the corresponding GRE T2* image, consistent with haemorrhage. Normal appearance of the internal cerebral veins, without signs of thrombosis
Vascular/ischaemic
Artery of Percheron infarction
In most individuals, the arterial supply of paramedian thalami is provided by pairs of thalamo-perforating arteries arising from P1 segments of both posterior cerebral arteries (PCAs). However, in a minority of cases, these perforating vessels stem from a single trunk arising from only one of the PCAs, known as the artery of Percheron. This variant is associated with an asymmetric fusion variant of the basilar tip.
An occlusion of this vessel results in bilateral thalamic infarctions with or without involvement of the rostral midbrain, and cardioembolic disease is considered the most frequent cause. The presenting acute symptoms include visual disturbances, confusion and coma [57, 58].
CT shows paramedian hypodense lesions of both thalami (Figs. 27a and 59), which are generally symmetric. While CT may be initially normal, as with other acute infarcts, MRI is much more sensitive demonstrating bright DWI signal with decreased diffusivity (Fig. 27 b and c) and T2 hyperintensity. The “midbrain V sign” is an additional characteristic imaging feature, which has been reported in the majority of patients. It consists of high signal intensity on FLAIR images that extends bilaterally along the midbrain surface at the interpeduncular cistern [58]. Volume loss with CSF-like density, signal intensity and diffusivity is found in chronic lesions (Fig. 28).

Artery of Percheron infarction. A 69-year-old female patient found unconscious. a Axial non-enhanced CT image shows subtle hypodense lesions (arrows) in both thalami. The lesions are bright on DWI (b) and dark on ADC map (c), consistent with reduced diffusivity of acute infarctions

Artery of Percheron infarction. Well-defined bilateral CSF-like hypodense lesions at centromedial thalami (arrowheads) represent chronic infarctions
Deep cerebral venous thrombosis
Patients with deep cerebral venous thrombosis (DCVT) tend to present with a few days’ history of headache and reduced consciousness. DCVT may be isolated or a part of a more widespread cerebral venous thrombosis, and it is associated with worse outcomes than dural venous thrombosis, due to the lack of a substantial collateral drainage pathways. Thrombosis of both internal cerebral veins leads to bilateral thalamic lesions, with variable involvement of the basal ganglia, internal capsule, midbrain and upper cerebellum [59].
On CT, there is a prominent hyperdensity of the occluded veins with hypodense and frequently haemorrhagic lesions of the thalami (Figs. 29 and 59) and, in some cases, basal ganglia. Depending on the presence of blood products and infarction, the MRI findings vary and are commonly heterogeneous, including T2 hyperintensity and hypointensity, low and high T1 signal (Fig. 30), as well as diffusion abnormality ranging from increased to decreased. In contrast to arterial infarcts, the observed signal abnormality is primarily due to venous congestion and resultant vasogenic oedema, rather than cytotoxic oedema. The lesions are characteristically bilateral, however strikingly asymmetric in some cases [60].

Deep cerebral venous thrombosis (DCVT). A 6-month-old boy with loss of consciousness. a Non-enhanced axial CT image demonstrates right greater than left bithalamic hyperdense lesion consistent with blood clot. There is mild mass effect and surrounding low attenuation (arrowheads) in the right thalamus. The blood clot appears contiguous with the hyperdensity (arrows) extending into the vein of Galen. b Reformatted midsagittal CT image confirms DCVT by depicting the prominent hyperdensity of the internal cerebral veins, vein of Galen (arrowhead) and straight sinus, as well as the inferior sagittal sinus (arrow)

DCVT. Term neonate with deep venous thrombosis. a There is bilateral swelling of the thalami with predominantly low signal intensity (arrowheads) on axial T1-weighted image. Left greater than right central hyperintense area is indicative of haemorrhage. Additionally, there is high signal intensity at the vein of Galen (arrow), suggestive of thrombosis. b MR venography reveals the absence of signal in the internal cerebral veins, vein of Galen and straight sinus, confirming DCVT
Dural arterio-venous fistula/vascular malformation
Non-haemorrhagic presentations of dural arterio-venous (AV) fistulas (dAVF) include dementia, which results from venous hypertension in the cortex or, less frequently, in bilateral thalami. The drainage of an dAVF into the deep venous system, primarily vein of Galen, causes bithalamic oedema and rapidly progressive dementia.
Mild swelling with decreased attenuation of the thalami may be seen on CT, while MRI demonstrates diffusely increased T2 signal intensity and diffusivity (high signal on ADC maps) of bilateral thalami with associated mild mass effect. There are no signs of DCVT and no haemorrhage, although microbleeds may be present on SW-MRI. The diagnosis is established by catheter angiogram (cerebral DSA), which can then be followed by endovascular embolisation treatment [61].
Neoplastic causes
Diffuse midline glioma
In the updated WHO 2016 classification of tumours of the central nervous system, a new entity, diffuse midline glioma with H3K27M mutant, has been added, referring to neoplasms previously designated as paediatric diffuse intrinsic pontine gliomas [62]. Further research has demonstrated that this mutation also occurs in other adult and paediatric midline and paramedial tumours, including thalamic gliomas [63]. Neoplastic lesions of the thalamus are relatively rare, predominantly affecting young adults and children. The deep localisation and the proximity to critical structures implies incomplete resection or non-resectability and poor prognosis. It has been recently found that bilateral thalamic gliomas, in contrast to their unilateral counterparts, frequently harbour mutations in EGFR oncogene, while H3K27M mutants are actually rare. Additionally, genomically tailored therapy (with tyrosine kinase inhibitors) showed encouraging results in a few paediatric patients with such bithalamic neoplasms [60, 64].
Both bilateral and unilateral thalamic gliomas show variable alterations in signal, diffusivity and contrast enhancement, but hyperintensity on T2-weighted and FLAIR images is typical. In contrast to other entities described in this review, a prominent mass effect is their characteristic feature (Figs. 31 and 59).

Bithalamic glioma. A 44-year-old male with dizziness, left face paraesthesia and weakness in the left side of body. a Axial T2-weighted image shows right greater than left expansion of the thalami with mildly heterogeneous increased signal intensity. b Corresponding ADC map reveals heterogeneous nature of the mass lesion with predominantly increased and peripherally reduced (arrows) diffusivity
Primary CNS lymphoma
Primary CNS lymphoma (PCNSL) most commonly arises from the deep brain structures and periventricular regions, usually contacting a CSF surface and with a tendency to spread along the subependymal lining. Medial thalami may be involved, occasionally in a bilateral symmetric fashion. In some cases, the lesions are not in contact with CSF surfaces and basal ganglia can also be affected. The typical imaging features are homogeneous intra-axial solitary or multiple well-defined masses, hyperdense (but of lower attenuation compared to acute blood clots) on non-enhanced CT (Fig. 32) and of low to isointense T2 signal, with marked surrounding vasogenic oedema. The lesions characteristically show dense homogeneous enhancement and very low diffusion with dark appearance on ADC maps (primarily due to high cellularity). Although the incidence of PCNSL had decreased slightly in the mid-1990s, concordantly with the decreasing rates of AIDS, it has been increasing in the elderly immunocompetent population [65,66,67]. When hyperdense homogeneous mass lesions with prominent surrounding oedema are seen along the ventricular surfaces on non-enhanced CT scan, the possibility of PCNSL should be raised.

Primary CNS lymphoma. A 46-year-old male with headache, collapse and vomiting. a, b Non-enhanced axial CT images show hyperdense lesions along the medial thalami (arrows) with adjacent low attenuation. An additional subependymal hyperdense lesion (arrowheads) is present along the frontal horn of the right lateral ventricle. There is also dilation of the ventricles with periventricular oedema and effacement of cortical sulci, consistent with acute hydrocephalus
Pathology involving both the basal ganglia and thalami
Inherited metabolic/genetic
Tubulinopathies or tubulin-related cortical dysgenesis
The tubulinopathies are a recently described heterogeneous group of brain malformations with a wide spectrum of clinical severity, which are caused by mutation of one of the seven genes encoding different isotypes of tubulin [68]. Tubulins and microtubule-associated proteins play an important role in cortical development. The diagnosis of a tubulinopathy is based on the presence of characteristic brain malformations on MRI and genetic testing.
Dysmorphic deep grey matter structures are the hallmark of the disease (Figs. 33, 34 and 59) and a consequence of abnormal white matter organisation in the internal capsules, such as hypoplasia of the anterior internal capsule. In most cases, the basal ganglia appear as large round structures in which the caudate, putamen and globus pallidus are indistinguishable, and the thalami are hypertrophic. In the microlissencephaly subtype, the basal ganglia are either not visible or severely hypoplastic. Furthermore, tubulinopathies are associated with brainstem hypoplasia and various malformations of the cerebellum, cerebral cortex and corpus callosum [69].

Tubulinopathy. A 10-year-old girl with mental retardation. There is a dysmorphic appearance of bilateral basal ganglia with fused and malformed caudate nucleus and putamen (arrowheads) on axial T1-weighted image. The thalami are hypoplastic (arrows). There is pachygyria in the insular regions along with the absence of the corpus callosum, Z-shaped brainstem and hypoplasia of the vermis and cerebellum (not shown)

Tubulinopathy. A 1-month-old girl with failure to thrive. The basal ganglia appear as large round structures with fusion of the caudate and putamen (arrowheads) on axial T2-weighted image. The thalami are slightly asymmetric
Neurofibromatosis type 1
Neurofibromatosis type 1 (NF-1) is the most common neurocutaneous syndrome/phakomatosis. The classic manifestations include café-au-lait macules, skinfold freckling, neurofibromas, optic pathway gliomas, hamartomas, Lisch nodules (iris hamartomas), characteristic bone lesions, such as sphenoid bone dysplasia, and arteriopathies, such as the Moya-Moya syndrome.
One of the predominant neuroimaging features are focal abnormal signal intensities (FASI), which are present in at least 60–80% of paediatric NF-1 patients. These are scattered ill-defined T2 hyperintense areas without mass effect in the basal ganglia, thalami, brainstem, cerebellum and occasionally cerebral hemispheres (Figs. 35, 36 and 59). FASIs can also show mild hyperintensity on T1-weighted images (Fig. 35b), while a minimal mass effect may occasionally be seen. These lesions are generally not visible on CT and very rarely enhance with gadolinium-based contrast agents. FASIs likely represent areas of vacuolar changes of myelin without evidence of demyelination. The importance of these findings is unknown and the causative relation with cognitive dysfunction is debated. It remains unclear why these changes frequently regress over time and even completely disappear, but can also reappear or temporarily increase in size in some cases [70,71,72]. The main differential diagnosis of FASIs is low-grade gliomas, which usually demonstrate a more substantial mass effect and, in some cases, contrast enhancement.

Focal abnormal signal intensities (FASIs) in NF-1. An 8-year-old boy with confirmed diagnosis of neurofibromatosis type 1 on genetic testing. a Axial FLAIR image demonstrates patchy bilateral focal abnormal signal intensities (FASIs) in the globi pallidi (arrowheads) and the posterior thalami (arrows). b On T1-weighted image, some of the lesions appear slightly hyperintense. c Axial T2-weighted image shows additional FASIs in the cerebellum (arrows)

FASIs in NF-1. Asymmetric ill-defined patchy hyperintensities consistent with FASIs (arrows) are seen in the basal ganglia and thalami on axial T2-weighted image
Primary familial brain calcification (Fahr disease)
Primary familial brain calcification (PFBC), formerly known as (familial) idiopathic basal ganglia calcification or Fahr disease, is a rare autosomal dominant neurodegenerative disease characterised by abnormal calcium deposits bilaterally in the basal ganglia and other brain regions. The disease usually presents as a combination of abnormal movements, cognitive and psychiatric manifestations, and is clinically indistinguishable from other adult-onset neurodegenerative disorders. Disease onset is usually in the fourth or sixth decade, although PFBC has been described in all age groups [73, 74].
In addition to the basal ganglia, the thalami (especially the posterolateral part), cerebellar dentate nuclei and subcortical white matter are also frequently affected on imaging studies and best seen on CT (Figs. 37 and 38). Less frequently, calcifications can be present in the cerebral and cerebellar cortex as well as brainstem. Confluent cerebral white matter abnormalities may also be associated. Secondary bilateral calcification is present in a variety of genetic, developmental, metabolic, infectious and other conditions. So the main goals in the evaluation of patients with movement disorder, cognitive impairment and psychiatric symptoms, in whom brain calcifications are detected on imaging, are to rule out metabolic disorders, primarily hypoparathyroidism as the most common cause of secondary calcifications, and to find out about the possible family history of the disease.

Primary familial brain calcification. A 30-year-old male with primary familial brain calcification (PFBC). Axial CT image demonstrates extensive calcifications in the basal ganglia, thalami and subcortical white matter. The putamina appear atrophic. There are also cerebellar calcifications in bilateral corpus medullare (not shown)

Primary familial brain calcification. A 57-year-old male with PFBC. Calcifications of the basal ganglia (arrows) and thalamus (arrowheads) are predominantly hyperintense on this sagittal T1-weighted image
Other conditions with similar intracranial calcification patterns include Cockayne syndrome and carbonic anhydrase deficiency type 2 [73, 75,76,77]. Labrune syndrome and Coats plus syndrome show dense rock-like calcifications; however, there are also white matter signal alterations, cysts and contrast enhancement [76].
Acquired metabolic/toxic
Ethylene glycol intoxication
Ethylene glycol (EG) is a colourless, odourless liquid and is the principal component of anti-freeze. EG ingestion is usually accidental. The life-threatening features are due to the metabolites of ethylene glycol in the same metabolic pathway as methanol. The accumulation of glycolic acid leads to metabolic acidosis and subsequent encephalopathy. The metabolic effects occur 12–24 h post-ingestion, comparable to methanol [77, 78].
Symmetric involvement of the central deep parts of the brain is the characteristic imaging finding of acute EG toxicity (within the first few days). A diffuse hypodensity of bilateral basal ganglia and thalami extending into the brainstem and adjacent cerebral hemispheres with mild mass effect is the typical pattern seen on the initial CT scan, which may, however, remain unremarkable. MRI reveals bilateral high T2 signal within the central basal portions of the brain (Figs. 39 and 59), primarily at the thalami and basal ganglia, hippocampi and amygdala, as well as the dorsal midbrain and pons. The signal abnormality may extend into the adjacent portions of the cerebellum, brainstem and insulae, along with a mild mass effect. Haemorrhagic lesions of bilateral globus pallidus or putamen, suggesting CO and methanol intoxication, have also been reported [77,78,79,80].

Ethylene glycol intoxication. A 25-year-old male found unconscious at home, after having his drink spiked with ethylene glycol (anti-freeze) the night before. The initial CT scan was unremarkable (not shown). a MRI 2 days later demonstrates bilateral symmetric swelling and hyperintensity in the basal ganglia and thalami on axial FLAIR image. The signal alteration is more prominent along the lateral margin of the putamina, with a suggestion of the lentiform fork sign on the right side (arrow). b Axial T2-weighted image through the pons shows increased signal and mild swelling of the dorsal brainstem (arrowheads) extending to the right middle cerebellar peduncle
Osmotic demyelination syndrome
Osmotic demyelination syndrome (ODS) is classically associated with a swift adjustment of hyponatraemia. However, it can result from a wide variety of electrolyte abnormalities and their correction, frequently encountered in chronically debilitated patients. ODS is histologically characterised by non-inflammatory destruction of myelin sheaths with sparing of the underlying axons. Features of the syndrome include quadriparesis and neurocognitive changes in the presence of typical lesions on brain MRI, representing central pontine myelinolysis (CPM) and extrapontine myelinolysis (EPM) [81].
There is, however, commonly a delay of the imaging features of several days or even weeks after the onset of symptoms, and reduced diffusivity on ADC maps may be the earliest finding. The hallmark of EPM is the striking symmetry of signal alterations, a feature of metabolic abnormalities, with T2 hyperintensity in the striatum, thalamus, external and extreme capsule (Fig. 40a). Corresponding hypodensities could be seen on CT (Fig. 41). The hippocampus, cerebellum and cerebral cortex may also be affected, while diffusivity depends on the scan timing. EPM may be seen without or in conjunction with CPM, which demonstrates the characteristic trident-shaped T2 hyperintense area with low T1 signal in the central pons (Fig. 40b) sparing the corticospinal tracts [81]. At least in some cases, EPM may precede CPM on imaging studies [82].

Osmotic demyelination syndrome (ODS)—extrapontine myelinolysis (EPM) and central pontine myelinolysis (CPM). A 46-year-old man with a background of alcohol abuse presented with hyponatraemia, which was corrected and followed by increasing neurological deficits, including quadriparesis and loss of consciousness. a Axial T2-weighted image at the basal ganglia level shows bilateral symmetric high signal intensity of the putamen and caudate head. There are also subtle hyperintensities in both thalami. b Axial T1-weighted image through the posterior fossa reveals a hypointense lesion in the pons with “trident” appearance (arrow), characteristic for CPM

ODS—extrapontine myelinolysis (EPM). A 7-year-old girl with a background of leukaemia presented with fever, vomiting and horizontal nystagmus. Hyponatraemia was established on admission and promptly adjusted. There is bilateral symmetric swelling and hypodensity of the basal ganglia centred at the caudate head and anterior lentiform nucleus (arrows) on non-enhanced axial CT image
Brain MRI changes with vigabatrin therapy
Vigabatrin, an effective antiepileptic medication for infantile spasms, is responsible for development of brain abnormalities on MRI in around 25% of treated infants. The characteristic imaging findings of symmetric high T2 signal and reduced diffusion are seen in the thalami, which may be accompanied with involvement of globi pallidi, brainstem tegmentum and cerebellar dentate nuclei (Figs. 42, 43 and 59) [83, 84]. These reversible and generally completely asymptomatic findings are associated with peak vigabatrin dosage. However, hyperkinetic movement disorders and life-threatening acute encephalopathy may develop in rare cases. The uncommon symptomatic MRI changes do not appear to be dose-dependent and could be associated with concomitant hormonal therapy. Vigabatrin-associated MRI abnormalities have not been found in older children and adults [84].

Brain MRI changes with vigabatrin therapy. A 50-week-old girl with infantile spasms treated with vigabatrin. a, b Axial T2-weighted images demonstrate bilateral symmetric hyperintensity of the thalami (arrowheads), basal ganglia (arrows), dentate nuclei (arrowheads) and brainstem (arrows). There is no associated mass effect. The findings were asymptomatic and resolved completely following discontinuation of vigabatrin. Courtesy of Kshitij (Kish) Mankand

Brain MRI changes with vigabatrin therapy. A 42-week-old girl treated with vigabatrin for epileptic encephalopathy. a There is decreased diffusivity of the thalami with low signal (arrows) on axial ADC map. b DWI at a lower level shows hyperintensity of the globi pallidi (arrows). Courtesy of Kshitij (Kish) Mankand
Endocrinological disorders related to parathyroid hormone disturbances
Cerebral calcifications are classified into three groups: physiological, idiopathic and secondary to calcium metabolism alterations. The most common aetiology of prominent bilateral intracranial calcifications are various types of hypoparathyroidism and pseudohypoparathyroidism, which can be caused by numerous disease processes. The observed bilateral symmetric calcifications in the globi pallidi, putamen and caudate nuclei on CT with correspondent hypointense signal on T2-weighted and SWI-MRI are sometimes referred to as Fahr syndrome. The thalami, subcortical white matter and cerebellar dentate nuclei are usually affected to a lesser extent (Fig. 44), followed by the cerebral cortex, hippocampus, subcortical and cerebellar white matter. The extent of calcification is variable, depending on the stage of the disease and duration of metabolic abnormalities [85, 86]. Associated findings in primary hypoparathyroidism are diffuse patchy osteolytic lesions in the skull, whereas plaque-like dural calcifications and vascular calcifications, most notably of carotid arteries, are encountered in secondary hyperparathyroidism.

Hyperparathyroidism. A 28-year-old male presented with new-onset tremor and hyperparathyroidism was established on laboratory testing. a, b CT demonstrates symmetric dense calcifications in the globi pallidi and dentate nuclei
Creutzfeldt–Jakob disease
Creutzfeldt–Jakob disease (CJD) is a rapidly progressive, fatal neurodegenerative disease typically characterised by dementia, ataxia, myoclonus and behavioural changes. CJD is a rare disorder with an annual incidence of 1–2 per million worldwide and is caused by accumulation of an abnormally shaped membrane-bound prion protein in neurons. It is subdivided into sporadic, familial and acquired forms. About 85–90% of cases are classified as sporadic CJD (sCJD), for which no aetiology can be identified. Genetic or familial CJD (fCJD), which is caused by 4 common and many other mutations in the prion protein gene, accounts for 10–15% of the cases. The acquired (infectious) form consists of iatrogenic CJD and variant CJD (vCJD, which originated as a zoonosis via consumption of contaminated beef) and comprises less than 1% of the cases [87].
MRI of the brain with DWI sequences is the most useful test with over 90% accuracy and should be obtained in all patients with a rapidly progressive neurological syndrome. The findings include hyperintensity on both FLAIR and DWI that is confined to the grey matter in the cortex (“cortical ribboning”), striatum (putamen and caudate nuclei), medial and/or posterior thalamus, or a combination of these areas, without any thickening or oedema (Figs. 45, 46 and 59). Decreased diffusivity with low signal on ADC maps is frequently present, but this is not very conspicuous and may change over time. In addition to bilateral findings, unilateral cortical and deep grey matter hyperintensity is frequently observed (Fig. 59) and represents a typical CJD pattern (in contrast to almost invariably symmetric lesions in metabolic and toxic disorders).

Creutzfeldt–Jakob disease (CJD). A 69-year-old male with a 5-month history of progressive generalised weakness, aphasia and cognitive decline and confirmed Creutzfeldt–Jakob disease (CJD). Axial DWI shows high signal intensity of bilateral striatum and dorsomedial thalami (“hockey stick” sign, arrowheads). There are also areas of cortical hyperintensity in the right operculum, as well as in bilateral insulae and cingulate gyri (arrows)

CJD. A 61-year-old female with rapidly progressive dementia, progressive motor and sensory impairment. CJD was confirmed. There is hyperintensity of bilateral basal ganglia, thalami and occipital cortices, along with partial temporal cortex involvement on axial DWI
The MRI criteria for sCJD are DWI signal abnormality from either at least two cortical regions or both the caudate nucleus and putamen [87, 88]. Cortical DWI hyperintensity also appears to be a presymptomatic marker, as the abnormalities on MRI have been found to antedate the onset of sCJD [89]. Bilateral symmetric posterior thalamic hyperintensity on FLAIR and DWI is known as the pulvinar sign (which along with involvement of dorsomedial thalami gives the “hockey stick” appearance) and is considered pathognomonic for vCJD, however only if the signal in the thalami is brighter than in the caudate and putamina [87].
Infectious/inflammatory
Acute disseminated or demyelinating encephalomyelitis
Acute disseminated encephalomyelitis aka ADEM is a usually transitory, monophasic and self-limiting immune-mediated inflammatory disease that can involve the brain and the spinal cord, typically affecting young children and adolescents. A preceding viral infection or less frequently immunisation is believed to trigger the inflammatory process based on cross-reactivity. However, many cases do not have a clear history of an infectious trigger, and an autoimmune aetiology is considered. In roughly 40% of the cases, antibodies to the myelin oligodendrocyte glycoprotein (MOG) are found [90]. Most patients recover completely with concomitant resolution of the imaging findings. In a minority of cases, the disease shows a multiphasic course or eventually progresses to multiple sclerosis [91].
On MRI, in addition to the white matter (and possibly spinal cord lesions), the basal ganglia and thalami are often bilaterally but asymmetrically involved with ill-defined T2 hyperintense lesions (Figs. 47 and 59) and variable heterogeneous contrast enhancement. Diffusivity of the lesions is usually increased (Fig. 47b); however, low diffusion with peripheral dark band on ADC maps is not uncommon and likely corresponds to advancing demyelination. The lesions may be hard to distinguish from other demyelinating diseases and infectious encephalitis (with selective grey matter involvement). ADEM associated with anti-MOG antibodies may also be combined with autoimmune encephalitis [92].

ADEM. A 4-year-old girl presented with behavioural change and seizure following upper respiratory infection. a Axial T2-weighted image at the basal ganglia level reveals focal high signal intensity of the dorsal right thalamus (arrowhead). There are also hyperintense white matter changes in the right temporal and left occipital lobes (arrows). b The lesions demonstrate increased diffusivity on the corresponding ADC map (arrows)
Acute haemorrhagic encephalomyelitis (AHEM, Hurst disease) is a rare and severe form characterised by rapidly progressive tumefactive white matter lesions and haemorrhagic foci, which are hypointense on GRE T2* or SW-MRI, with variable basal ganglia and thalamic involvement. AHEM with thalamic involvement may occasionally have an appearance very similar to ANE.
Viral encephalitis
Viruses have a propensity to affect grey matter structures, leading to a fairly common, usually asymmetrical, high T2 signal intensity on MRI and hypodensity on CT in bilateral thalami and/or basal ganglia. Consequently, viral infection should always be taken into consideration when such lesions are found in an appropriate clinical setting [93]. Some of the viruses characteristically lead to a specific localisation and/or imaging appearance of lesions in the CNS; however, they may also present with non-specific findings, while other infections and non-infectious diseases may simulate their typical presentation [93].
Arboviruses (arthropod borne viruses) include three different groups of infectious agents that are transmitted by insects, most commonly mosquitoes. Flaviviridae is the most prominent group that includes agents causing Japanese and West Nile, as well as tick-borne encephalitis. The characteristic imaging findings include bilateral focal patchy thalamic lesions that are of low attenuation on CT and of high T2 signal intensity, typically bright on DWI (usually with reduced diffusivity on ADC maps, at least in some parts). The basal ganglia and brainstem are frequently affected, and cortical abnormalities may also be found (Figs. 48 and 59). There is some mass effect (with expansion of the anatomic structures, not a focal mass) in larger lesions and haemorrhages may be seen, while contrast enhancement is variable. The spinal cord and cauda equina can also be involved [94, 95].

West Nile viral encephalitis. A 71-year-old male with severe headache, high fever, disorientation, tremors and abnormal movements due to West Nile virus encephalitis. Coronal FLAIR image shows hyperintensity of the right thalamus (arrow), right periopercular fronto-insulo-temporal region, bilateral hippocampi and left uncus
Epstein–Barr virus is one of the members of herpesvirus family that can cause neurologic diseases. Most cases of EBV encephalitis occur in children as a rare complication of infectious mononucleosis [96]. EBV encephalitis is frequently associated with abnormalities in the deep grey matter, and bilateral involvement of the basal ganglia and thalami is characteristic, frequently with associated cortical lesions (Fig. 49) [97].

EBV viral encephalitis. A 6-year-old boy with Epstein–Barr virus encephalitis. There is a hyperintense lesion with mild mass effect involving the left caudate head and ventral putamen (arrow) on axial FLAIR image. Furthermore, there are cortical lesions of high signal intensity in bilateral cingulate gyri and right insula (arrowheads)
Cerebral toxoplasmosis
Cerebral toxoplasmosis, caused by the protozoan parasite Toxoplasma gondii, is the most common opportunistic CNS infection in AIDS patients, who are especially susceptible to reactivation of a latent infection when their CD4 count falls below 100 cells/μL [98].
The lesions are typically located in the deep grey matter and at corticomedullary junction, appearing as multiple (or solitary) areas of low attenuation on unenhanced CT and exhibiting nodular or ring enhancement on post-contrast CT and T1-weighted MR images. Ring-enhancing lesions can show an internal eccentric nodule, referred to as the “eccentric target sign” (Figs. 50 and 59), which is highly specific for toxoplasmosis, but seen in less than 30% of the cases. Another specific MRI feature is the “concentric target sign” in T2-weighted and FLAIR images with alternating concentric layers of hypo- and hyperintensities (a hypointense core, an intermediate hyperintense area and a peripheral hypointense zone), surrounded by high signal of vasogenic oedema (Figs. 51 and 59).

Cerebral toxoplasmosis. A 32-year-old man with known HIV infection and a 5-day history of increasing confusion, headache and fever. Axial post-contrast T1-weighted image demonstrates an oval enhancing lesion at the left basal ganglia with surrounding vasogenic oedema and prominent mass effect. The enhancement is peripheral with an internal nodular extension (arrow), representing the “eccentric target” sign of toxoplasmosis

Cerebral toxoplasmosis. A 38-year-old woman presenting with fever, headache, blurred vision, seizures and left-sided weakness was found to be HIV positive. a There are at least three areas of vasogenic oedema in this axial FLAIR image. Within the oedema of the right hemisphere, there is a round retrolenticular lesion with internal layers—concentric alternating hypointense and hyperintense areas (arrow). This characteristic appearance represents the “concentric target” sign of toxoplasmosis
The lesions of toxoplasmosis demonstrate decreased rCBV on perfusion imaging (which is typical for inflammatory and infectious lesions) and usually central high signal of increased diffusivity on ADC maps, in contrast to PCNSL, which is the main differential diagnosis in HIV-infected patients [98, 99].
Vascular/ischaemic
Hypoxic–ischaemic encephalopathy and status marmoratus
The pattern of brain injury in global hypoxia depends on the level of brain maturation at the time of insult and the severity and duration of the event. Anoxic injury may range from mild to moderate and severe. It is the severe injury that affects the central grey matter structures in infants, young children and adults.
Perinatal asphyxia is the most important cause of HIE and the damaged areas parallel the myelinating regions. Severe HIE in preterm neonates results in lesions of the thalami, dorsal brainstem and anterior vermis. Conversely, in term neonates and infants up to 1 year, the globi pallidi, posterior putamina, ventrolateral thalami, hippocampi, dorsal brainstem and sensorimotor cortex are most affected. When bilateral basal ganglia lesions are seen in a neonate and there is little evidence for perinatal hypoxia, inborn errors of metabolism, such as sulphite oxidase deficiency, should be considered as a differential diagnosis [100]. HIE in older children and adults causes diffuse injury to the grey matter with symmetric involvement of the basal ganglia, thalami and cortex (Figs. 52 and 59).

Hypoxic–ischaemic encephalopathy (HIE). A 44-year-old female following resuscitation after ventricular fibrillation and cardiac arrest. a There is bilateral hyperintensity of the striatum and predominantly posterior cortical grey matter, along with focal thalamic lesions (arrows) on axial DWI. The lesions are so diffuse and widespread that the image might even appear normal (with a wider window setting), except for the fact that the globi pallidi (*) and most of the thalami are spared and not as bright. b There is reduced diffusivity of the lesions on corresponding ADC map (arrows), consistent with cytotoxic oedema. In contrast to most other clinical settings, the abnormally low ADC signal is actually more conspicuous than the DWI hyperintensity
The affected areas show decreased diffusivity followed by T2 hyperintensity on MRI and hypodensity on CT. Diffusion MR imaging in perinatal HIE reliably shows hyperintense signal on DWI and reduced diffusivity on ADC maps after 24 h (Fig. 53a), which peaks at 3–5 days after the hypoxic event, which represents the optimal timeframe to scan the neonates. Afterwards, pseudonormalisation of diffusivity occurs on ADC maps. T2 hyperintensity of the basal ganglia and thalami is generally present after 2 days and slowly disappears over time. By the third day, T1 shortening will be seen in the injured areas (Fig. 53b) and remain for many months [100, 101]. This leads to a characteristic reversal of normal T1 hyperintensity in the myelinated posterior limb of the internal capsule (PLIC) compared to the adjacent deep grey matter structures (Figs. 54 and 59) in term neonates (the basal ganglia and thalami become brighter than PLIC on T1-weighted images).

HIE. Term neonate with peripartal asphyxia and convulsions. a There is bilateral decreased diffusivity with low ADC values compatible with cytotoxic oedema in the globi pallidi and ventrolateral thalami (arrows). b There is absence of the normal high signal in the myelinated PLIC (arrows), which should be present in a term neonate on axial T1-weighted image. This is known as the “absent posterior limb” sign

HIE. A 5-day-old term neonate with severe perinatal asphyxia. a Axial T1-weighted and b T2-weighted images demonstrate bilateral symmetric signal alteration in the ventrolateral thalami, globi pallidi and posterior putamina, which are hyperintense (arrows) and hypointense, respectively. The findings are compatible with HIE. Note the bilateral high T1 signal of myelin in the posterior limb of the internal capsule (PLIC), which is normal in neonates after 37 weeks of gestational age
The diffusion abnormalities are essentially always present on MRI scan following anoxic injury in adults (most commonly following cardiac arrest) and older children. Although the clinical context of HIE is mostly clear, an alternative diagnosis or combined pathophysiology may be considered in the unconscious patient of unknown aetiology, such as toxic encephalopathy or metabolic acidosis.
Status marmoratus or “état marbré” is a rare manifestation of the chronic features of profound perinatal HIE in full-term neonates. The initial ischaemic damage evolves to gliosis and is mixed with an abnormal or excessive myelination in the second part of the first year of life, causing a marbled appearance of the basal ganglia and thalami [102]. On MRI, the hypermyelination is most apparent in the putamen. There is abnormal T2 hyperintensity throughout the basal ganglia and thalami, often combined with severe cerebral atrophy.
Hypertensive haematoma
Hypertensive haemorrhages typically occur in areas supplied by penetrating vessels with the most common locations being the central grey matter, primarily the putamen (around 65%) and thalamus (about 20%), followed by the pons and medial cerebellum. The imaging modality in the acute setting is usually CT, which reveals hyperdense lesion with attenuation values of blood clot in these characteristic locations and is highly suggestive of the diagnosis. Thin peripheral contrast enhancement may be observed around the haematoma [103].
Simultaneous bilateral hypertensive basal ganglia and/or thalamic haemorrhages may rarely be encountered in the acute setting (Figs. 55 and 56b), usually asymmetric [104, 105]. The hypertensive haematomas typically dissect the brain without a considerable amount of associated tissue necrosis, so that chronic lesions are transformed into slit-like cavities with peripheral low T2 signal due to haemosiderin staining (Fig. 56b), which is accentuated on SW-MRI or GRE T2* images [103]. In contrast to acute lesions, chronic bilateral hypertensive haematomas are not an uncommon finding in deep grey matter.

Hypertensive haematomas. A 75-year-old female presenting with left hemiparesis and Glasgow coma score (GCS) 6. Non-enhanced axial CT image shows right greater than left hyperdense lesions (arrows) at the lentiform nucleus and posterior limb of the internal capsule with a thin rim of low attenuation, consistent with acute hypertensive haematomas

Hypertensive haematomas. A 59-year-old male with sudden loss of consciousness. a There are left greater than right acute hypertensive haemorrhages centred at putamina on this non-enhanced axial CT image. There are also adjacent surrounding areas of low attenuation, consistent with extruded serum and oedema around the blood clot, and a prominent mass effect on the left. b Follow-up T2-weighted image 2 years later reveals corresponding slit-like lesions with peripheral hypointensity (arrowheads) representing haemosiderin staining
Posterior reversible encephalopathy syndrome
Posterior reversible encephalopathy syndrome (PRES) is classically and commonly triggered by an acute hypertensive state. However, multiple other conditions may predispose to PRES. Damage to the vascular endothelium with subsequent disruption of cerebral vascular autoregulation is the cornerstone of the pathophysiology, resulting in vasogenic oedema.
Typical MRI findings include patchy, near-symmetrical T2 hyperintense areas in the subcortical regions of the posterior cerebral hemispheres (Fig. 57a). The overlying cortex is frequently involved, and there may be some patchy contrast enhancement. The diffusivity is typically increased with high signal on ADC maps. In some cases, however, there are areas with decreased diffusivity, indicating irreversible infarction (Fig. 57b). Other parts of the brain, including anterior frontal lobes and infratentorial structures, may also be affected [106, 107].

Posterior reversible encephalopathy syndrome (PRES). A 2-month-old boy with PRES. a Axial T2-weighted image reveals bilateral confluent hyperintense areas in the cortico-subcortical parieto-occipital regions (arrows) along with right greater than left involvement of the globi pallidi (arrowheads). b The lesions are predominantly showing increased diffusivity with high signal on the corresponding ADC map representing vasogenic oedema. There are, however, a few small cortical areas of reduced diffusivity (arrows) indicative of cytotoxic oedema
A recent review of over 550 reported cases found that the atypical locations of PRES are most commonly in the cerebellum, brainstem and deep white matter, along with the basal ganglia (in 24.3%) and thalamus (in 20.5%). The common symptoms of such PRES include headaches, altered mental status, seizures, visual disturbances, nausea or vomiting and focal neurological deficits. The frequent predisposing factors include hypertension, renal diseases, immunosuppressant drugs and chemotherapy/chemoradiotherapy [107].
In the classic presentation of PRES, the basal ganglia are involved in only 10–20% of patients (Figs. 57 and 59). However, a “central variant” PRES shows predominant involvement of the brainstem and the basal ganglia with variable involvement of the thalami and periventricular white matter, while the parieto-occipital and posterior frontal cortico-subcortical regions are spared (Fig. 58). Differential diagnosis can be very broad because of the atypical imaging features and could include osmotic demyelination, encephalitis and demyelination [106].

PRES. A 68-year-old male presenting with mental status change, headache, ataxia and dizziness. High blood pressure on exam. a There are scattered hyperintense lesions in bilateral caudate heads, lentiform nuclei (arrowheads), thalami (arrows) and periventricular white matter on axial FLAIR image. b Corresponding ADC map shows mildly increased diffusivity of the lesions, best seen in the thalami (arrows)
Conclusion
A systematic approach combining imaging, clinical and demographic data is necessary when encountering lesions of the central grey matter on scans of the brain. Knowledge of the exact localisation of the abnormalities and the characteristic findings on various MR sequences, and occasionally on CT, allows for narrowing down the differential considerations and even establishing the final diagnosis in some cases. In addition to the findings in the basal ganglia and thalami, answers to 3 main questions are very helpful when such lesions are found on imaging studies:
-
1
Imaging—Is there involvement of additional areas of the brain? More specifically, of the following three:
-
(a)
Brainstem
-
(b)
Cortex
-
(c)
White matter
-
(a)
-
2
Demographics—What is the patient’s age?
-
3
Clinical—Acute or chronic presentation?
A summary imaging overview showing the most characteristic disease patterns involving the basal ganglia and thalami discussed in this paper is provided in Fig. 59. The various disease entities are furthermore summarised in Table 1.

Summary overview of most pathologies listed in this review paper. Predominantly involving the basal ganglia. Leigh Sy—BL symmetric BG and brainstem (+/− thalami); glutaric aciduria type 1—BL BG and WM + wide Sylvian fissures; WD—BL symmetric striatum on T2w (+/− brainstem and thalami), GP on T1w; HD—BL atrophy of striatum—“boxed-out” frontal horns; PKAN—BL GP “eye of the tiger” sign; CO poisoning—BL focal GP lesions; kernicterus—BL GP (+ subthalamic nucleus); chronic HE/Mn/Gd deposition—BL GP on T1w; methanol poisoning—BL putamen (+ haemorrhage) and frontal WM; uremic encephalopathy/metabolic acidosis—BL lentiform fork sign; hypoglycaemia—BL BG and diffuse cortical GM on DWI; hyperglycaemia—unilateral striatum on T1w and CT; autoimmune encephalitis—BL striatum (+/− hippocampi and other areas); cryptococcosis—scattered focal BL BG lesions; germinoma—UL or BL on T2*w, hemiatrophy. Predominantly involving the thalami. Gangliosidosis—T1 bright and T2 dark thalami; toluene toxicity—T2 dark thalami and hyperintense WM, atrophy; WE—BL paramedial thalami (+ periaqueductal GM and mammillary bodies); acute hyperammonaemia—BL GM of insula, cingulate and thalami; ANE—BL concentric haemorrhagic thalamic lesions; artery of Percheron infarction—BL focal thalamic infarcts (+/− midbrain); DCVT—thalamic swelling with haemorrhage and venous thrombosis; bithalamic glioma—prominent mass effect. Affecting the basal ganglia and thalami. Tubulinopathy—fused malformed BG + other findings; NF-1 FASI—scattered patchy ill-defined; EG poisoning—BL BG and thalami; vigabatrin-associated MRI changes—BL thalami and BG (+ brainstem and dentate nuclei); CJD—BL or UL striatum and cortex (+/− thalami—“hockey stick” sign) on DWI; ADEM—asymmetric central GM and WM; viral encephalitis—asymmetric central and cortical GM; toxoplasmosis—eccentric and concentric target sign; HIE adult—symmetric central and cortical GM on ADC; HIE neonate—absent PLIC, T1 bright central GM; PRES—BL WM and GM oedema. ADEM, acute disseminated encephalomyelitis; AIE, autoimmune encephalitis; AoP, artery of Percheron; ANE, acute necrotising encephalitis; BG, basal ganglia; BL, bilateral; CJD, Creutzfeldt–Jakob disease; CO, carbon monoxide; DCVT, deep cerebral venous thrombosis; FASIs, focal abnormal signal intensities; GA, glutaric aciduria; GM, grey matter; GP, globus pallidus; HD, Huntington disease; HIE, hypoxic–ischaemic encephalopathy; PKAN, pantothenate kinase-associated neurodegeneration; PRES, posterior reversible encephalopathy syndrome; UL, unilateral; WD, Wilson disease; WE, Wernicke encephalopathy; WM, white matter
Author declarations
Idea for this review: Zoran Rumboldt and Luc van den Hauwe.
Literature search and data analysis: Sofie Van Cauter, Mariasavina Severino, Rosamaria Ammendola, Brecht Van Berkel, Hrvoje Vavro, Luc van den Hauwe, and Zoran Rumboldt.
Drafting of the article: Sofie Van Cauter, Mariasavina Severino, and Zoran Rumboldt.
Critical revision of the manuscript: Sofie Van Cauter, Mariasavina Severino, Rosamaria Ammendola, Brecht Van Berkel, Hrvoje Vavro, Luc van den Hauwe, and Zoran Rumboldt.
References
Hoch MJ, Bruno MT, Faustin A, Cruz N, Mogilner AY, Crandall L, Wisniewski T, Devinsky O, Shepherd TM (2019) 3T MRI whole-brain microscopy discrimination of subcortical anatomy, part 2: basal forebrain. AJNR Am J Neuroradiol 40(7):1095–1105. https://doi.org/10.3174/ajnr.A6088
ten Donkelaar HJ, Lammens M, Akira H (2014) Clinical neuroembryology: development and developmental disorders of the human central nervous system. Springer, New York
Dixon AKB, David J.; Ellis, Harold; Logan Bari M. (2015) Brain: selected images. In: Human sectional anatomy: atlas of body sections, CT and MRI images, fourth edition. CRC
Adams LC, Bressem K, Boker SM, Bender YY, Norenberg D, Hamm B, Makowski MR (2017) Diagnostic performance of susceptibility-weighted magnetic resonance imaging for the detection of calcifications: a systematic review and meta-analysis. Sci Rep 7(1):15506. https://doi.org/10.1038/s41598-017-15860-1
Henkelman RM, Watts JF, Kucharczyk W (1991) High signal intensity in MR images of calcified brain tissue. Radiology 179(1):199–206. https://doi.org/10.1148/radiology.179.1.1848714
Tullo S, Patel R, Devenyi GA, Salaciak A, Bedford SA, Farzin S, Wlodarski N, Tardif CL, Group P-AR, JCS B, Chakravarty MM (2019) MR-based age-related effects on the striatum, globus pallidus, and thalamus in healthy individuals across the adult lifespan. Hum Brain Mapp 40(18):5269–5288. https://doi.org/10.1002/hbm.24771
Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJ, Mayatepek E, Morava E, Distelmaier F (2014) A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry 85(3):257–265. https://doi.org/10.1136/jnnp-2012-304426
Arii J, Tanabe Y (2000) Leigh syndrome: serial MR imaging and clinical follow-up. AJNR Am J Neuroradiol 21(8):1502–1509
Kolker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, Burlina AP, Dixon M, Duran M, Garcia Cazorla A, Goodman SI, Koeller DM, Kyllerman M, Muhlhausen C, Muller E, Okun JG, Wilcken B, Hoffmann GF, Burgard P (2011) Diagnosis and management of glutaric aciduria type I--revised recommendations. J Inherit Metab Dis 34(3):677–694. https://doi.org/10.1007/s10545-011-9289-5
Nunes J, Loureiro S, Carvalho S, Pais RP, Alfaiate C, Faria A, Garcia P, Diogo L (2013) Brain MRI findings as an important diagnostic clue in glutaric aciduria type 1. Neuroradiol J 26(2):155–161. https://doi.org/10.1177/197140091302600204
Christian CW, States LJ (2017) Medical mimics of child abuse. AJR Am J Roentgenol 208(5):982–990. https://doi.org/10.2214/AJR.16.17450
Ranjan A, Kalita J, Kumar S, Bhoi SK, Misra UK (2015) A study of MRI changes in Wilson disease and its correlation with clinical features and outcome. Clin Neurol Neurosurg 138:31–36. https://doi.org/10.1016/j.clineuro.2015.07.013
Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW, Seo JK, Yeon KM (2006) MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. AJNR Am J Neuroradiol 27(6):1373–1378
Sinha S, Taly AB, Ravishankar S, Prashanth LK, Venugopal KS, Arunodaya GR, Vasudev MK, Swamy HS (2006) Wilson’s disease: cranial MRI observations and clinical correlation. Neuroradiology 48(9):613–621. https://doi.org/10.1007/s00234-006-0101-4
Scahill RI, Andre R, Tabrizi SJ, Aylward EH (2017) Structural imaging in premanifest and manifest Huntington disease. Handb Clin Neurol 144:247–261. https://doi.org/10.1016/B978-0-12-801893-4.00020-1
Hobbs NZ, Barnes J, Frost C, Henley SM, Wild EJ, Macdonald K, Barker RA, Scahill RI, Fox NC, Tabrizi SJ (2010) Onset and progression of pathologic atrophy in Huntington disease: a longitudinal MR imaging study. AJNR Am J Neuroradiol 31(6):1036–1041. https://doi.org/10.3174/ajnr.A2018
Amaral LL, Gaddikeri S, Chapman PR, Roy R, Gaddikeri RS, Marussi VH, Bag AK (2015) Neurodegeneration with brain iron accumulation: clinicoradiological approach to diagnosis. J Neuroimaging 25(4):539–551. https://doi.org/10.1111/jon.12195
McNeill A, Birchall D, Hayflick SJ, Gregory A, Schenk JF, Zimmerman EA, Shang H, Miyajima H, Chinnery PF (2008) T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology 70(18):1614–1619. https://doi.org/10.1212/01.wnl.0000310985.40011.d6
Beppu T (2013) The role of MR imaging in assessment of brain damage from carbon monoxide poisoning: a review of the literature. Am J Neuroradiol 35(4):625–631. https://doi.org/10.3174/ajnr.A3489
Jeon SB, Sohn CH, Seo DW, Oh BJ, Lim KS, Kang DW, Kim WY (2018) Acute brain lesions on magnetic resonance imaging and delayed neurological sequelae in carbon monoxide poisoning. JAMA Neurol 75(4):436–443. https://doi.org/10.1001/jamaneurol.2017.4618
Wisnowski JL, Panigrahy A, Painter MJ, Watchko JF (2014) Magnetic resonance imaging of bilirubin encephalopathy: current limitations and future promise. Semin Perinatol 38(7):422–428. https://doi.org/10.1053/j.semperi.2014.08.005
Rovira A, Alonso J, Cordoba J (2008) MR imaging findings in hepatic encephalopathy. AJNR Am J Neuroradiol 29(9):1612–1621. https://doi.org/10.3174/ajnr.A1139
Choi JW, Moon WJ (2019) Gadolinium deposition in the brain: current updates. Korean J Radiol 20(1):134–147. https://doi.org/10.3348/kjr.2018.0356
Gulani V, Calamante F, Shellock FG, Kanal E, Reeder SB, International Society for Magnetic Resonance in M (2017) Gadolinium deposition in the brain: summary of evidence and recommendations. Lancet Neurol 16(7):564–570. https://doi.org/10.1016/S1474-4422(17)30158-8
Zakharov S, Kotikova K, Vaneckova M, Seidl Z, Nurieva O, Navratil T, Caganova B, Pelclova D (2016) Acute methanol poisoning: prevalence and predisposing factors of haemorrhagic and non-haemorrhagic brain lesions. Basic Clin Pharmacol Toxicol 119(2):228–238. https://doi.org/10.1111/bcpt.12559
Taheri MS, Moghaddam HH, Moharamzad Y, Dadgari S, Nahvi V (2010) The value of brain CT findings in acute methanol toxicity. Eur J Radiol 73(2):211–214. https://doi.org/10.1016/j.ejrad.2008.11.006
Kumar G, Goyal MK (2010) Lentiform fork sign: a unique MRI picture. Is metabolic acidosis responsible? Clin Neurol Neurosurg 112(9):805–812. https://doi.org/10.1016/j.clineuro.2010.06.006
Kim DM, Lee IH, Song CJ (2016) Uremic encephalopathy: MR imaging findings and clinical correlation. AJNR Am J Neuroradiol 37(9):1604–1609. https://doi.org/10.3174/ajnr.A4776
Bathla G, Policeni B, Agarwal A (2014) Neuroimaging in patients with abnormal blood glucose levels. AJNR Am J Neuroradiol 35(5):833–840. https://doi.org/10.3174/ajnr.A3486
Wong DS, Poskitt KJ, Chau V, Miller SP, Roland E, Hill A, Tam EW (2013) Brain injury patterns in hypoglycemia in neonatal encephalopathy. AJNR Am J Neuroradiol 34(7):1456–1461. https://doi.org/10.3174/ajnr.A3423
Aquino JH, Spitz M, Pereira JS (2015) Hemichorea-hemiballismus as the first sign of type 1b diabetes during adolescence and its recurrence in the setting of infection. J Child Neurol 30(10):1362–1365. https://doi.org/10.1177/0883073814553972
Oh SH, Lee KY, Im JH, Lee MS (2002) Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a meta-analysis of 53 cases including four present cases. J Neurol Sci 200(1–2):57–62. https://doi.org/10.1016/s0022-510x(02)00133-8
Kelley BP, Patel SC, Marin HL, Corrigan JJ, Mitsias PD, Griffith B (2017) Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol 38(6):1070–1078. https://doi.org/10.3174/ajnr.A5086
Crespo-Burillo JA, Hernando-Quintana N, Ruiz-Palomino P, Martin-Martinez J (2015) Chorea secondary to striatal encephalitis due to anti-CV2/CRMP5 antibodies. Case description and review of the literature. Neurologia 30(7):451–453. https://doi.org/10.1016/j.nrl.2013.10.007
da Rocha AJ, Nunes RH, Maia AC Jr, do Amaral LL (2015) Recognizing autoimmune-mediated encephalitis in the differential diagnosis of limbic disorders. AJNR Am J Neuroradiol 36(12):2196–2205. https://doi.org/10.3174/ajnr.A4408
Charlier C, Dromer F, Leveque C, Chartier L, Cordoliani YS, Fontanet A, Launay O, Lortholary O, French Cryptococcosis Study G (2008) Cryptococcal neuroradiological lesions correlate with severity during cryptococcal meningoencephalitis in HIV-positive patients in the HAART era. PLoS One 3(4):e1950. https://doi.org/10.1371/journal.pone.0001950
Corti M, Villafane MF, Negroni R, Arechavala A, Maiolo E (2008) Magnetic resonance imaging findings in AIDS patients with central nervous system cryptococcosis. Rev Iberoam Micol 25(4):211–214. https://doi.org/10.1016/s1130-1406(08)70051-2
Zhong Y, Zhou Z, Fang X, Peng F, Zhang W (2017) Magnetic resonance imaging study of cryptococcal neuroradiological lesions in HIV-negative cryptococcal meningitis. Eur J Clin Microbiol Infect Dis 36(8):1367–1372. https://doi.org/10.1007/s10096-017-2941-8
Lou X, Ma L, Wang FL, Tang ZP, Huang H, Cai YQ, Wong EH (2009) Susceptibility-weighted imaging in the diagnosis of early basal ganglia germinoma. AJNR Am J Neuroradiol 30(9):1694–1699. https://doi.org/10.3174/ajnr.A1696
Phi JH, Cho BK, Kim SK, Paeng JC, Kim IO, Kim IH, Kim DG, Jung HW, Kim JE, Wang KC (2010) Germinomas in the basal ganglia: magnetic resonance imaging classification and the prognosis. J Neuro-Oncol 99(2):227–236. https://doi.org/10.1007/s11060-010-0119-7
Patterson MC (2013) Gangliosidoses. Handb Clin Neurol 113:1707–1708. https://doi.org/10.1016/B978-0-444-59565-2.00039-3
Gururaj A, Sztriha L, Hertecant J, Johansen JG, Georgiou T, Campos Y, Drousiotou A, d’Azzo A (2005) Magnetic resonance imaging findings and novel mutations in GM1 gangliosidosis. J Child Neurol 20(1):57–60. https://doi.org/10.1177/08830738050200010901
Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, van Wieringen WN, Barkhof F, Wolf NI, van der Knaap MS (2010) Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 133(10):2971–2982. https://doi.org/10.1093/brain/awq257
Autti T, Joensuu R, Aberg L (2007) Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology 49(7):571–578. https://doi.org/10.1007/s00234-007-0220-6
Kumar D, Ramanathan S, Khanna M, Palaniappan Y (2014) Bithalamic T2 hypointensity: a diagnostic clue for Sandhoff’s disease. Neurol India 62(4):481–482. https://doi.org/10.4103/0028-3886.141311
Cocozza S, Russo C, Pontillo G, Pisani A, Brunetti A (2018) Neuroimaging in Fabry disease: current knowledge and future directions. Insights Imaging 9(6):1077–1088. https://doi.org/10.1007/s13244-018-0664-8
Aydin K, Sencer S, Demir T, Ogel K, Tunaci A, Minareci O (2002) Cranial MR findings in chronic toluene abuse by inhalation. AJNR Am J Neuroradiol 23(7):1173–1179
Zuccoli G, Santa Cruz D, Bertolini M, Rovira A, Gallucci M, Carollo C, Pipitone N (2009) MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. AJNR Am J Neuroradiol 30(1):171–176. https://doi.org/10.3174/ajnr.A1280
Manzo G, De Gennaro A, Cozzolino A, Serino A, Fenza G, Manto A (2014) MR imaging findings in alcoholic and nonalcoholic acute Wernicke’s encephalopathy: a review. Biomed Res Int 2014:503596–503512. https://doi.org/10.1155/2014/503596
Mach JC, Russell J (2020) A combined case of Wernicke and metronidazole induced encephalopathy? Overlapping pathophysiologic pathways and MR imaging features. Clin Neurol Neurosurg 196:106034. https://doi.org/10.1016/j.clineuro.2020.106034
Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH (2007) MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol 28(9):1652–1658. https://doi.org/10.3174/ajnr.A0655
U-King-Im JM, Yu E, Bartlett E, Soobrah R, Kucharczyk W (2011) Acute hyperammonemic encephalopathy in adults: imaging findings. AJNR Am J Neuroradiol 32(2):413–418. https://doi.org/10.3174/ajnr.A2290
Reis E, Coolen T, Lolli V (2020) MRI findings in acute hyperammonemic encephalopathy: three cases of different etiologies: teaching point: to recognize MRI findings in acute hyperammonemic encephalopathy. J Belg Soc Radiol 104(1):9. https://doi.org/10.5334/jbsr.2017
Wu X, Wu W, Pan W, Wu L, Liu K, Zhang HL (2015) Acute necrotizing encephalopathy: an underrecognized clinicoradiologic disorder. Mediat Inflamm 2015:792578–792510. https://doi.org/10.1155/2015/792578
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B (2020) COVID-19-associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology 296(2):E119–E120. https://doi.org/10.1148/radiol.2020201187
Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, Sultan M, Easton A, Breen G, Zandi M, Coles JP, Manji H, Al-Shahi Salman R, Menon DK, Nicholson TR, Benjamin LA, Carson A, Smith C, Turner MR, Solomon T, Kneen R, Pett SL, Galea I, Thomas RH, Michael BD, CoroNerve Study G (2020) Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry doi:https://doi.org/10.1016/S2215-0366(20)30287-X
Arauz A, Patino-Rodriguez HM, Vargas-Gonzalez JC, Arguelles-Morales N, Silos H, Ruiz-Franco A, Ochoa MA (2014) Clinical spectrum of artery of Percheron infarct: clinical-radiological correlations. J Stroke Cerebrovasc Dis 23(5):1083–1088. https://doi.org/10.1016/j.jstrokecerebrovasdis.2013.09.011
Lazzaro NA, Wright B, Castillo M, Fischbein NJ, Glastonbury CM, Hildenbrand PG, Wiggins RH, Quigley EP, Osborn AG (2010) Artery of Percheron infarction: imaging patterns and clinical spectrum. AJNR Am J Neuroradiol 31(7):1283–1289. https://doi.org/10.3174/ajnr.A2044
Linn J, Pfefferkorn T, Ivanicova K, Muller-Schunk S, Hartz S, Wiesmann M, Dichgans M, Bruckmann H (2009) Noncontrast CT in deep cerebral venous thrombosis and sinus thrombosis: comparison of its diagnostic value for both entities. AJNR Am J Neuroradiol 30(4):728–735. https://doi.org/10.3174/ajnr.A1451
Chen XY, Wang Q, Wang X, Wong KS (2017) Clinical features of thalamic stroke. Curr Treat Options Neurol 19(2):5. https://doi.org/10.1007/s11940-017-0441-x
Holekamp TF, Mollman ME, Murphy RK, Kolar GR, Kramer NM, Derdeyn CP, Moran CJ, Perrin RJ, Rich KM, Lanzino G, Zipfel GJ (2016) Dural arteriovenous fistula-induced thalamic dementia: report of 4 cases. J Neurosurg 124(6):1752–1765. https://doi.org/10.3171/2015.5.JNS15473
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820. https://doi.org/10.1007/s00401-016-1545-1
Kleinschmidt-DeMasters BK, Mulcahy Levy JM (2018) H3 K27M-mutant gliomas in adults vs. children share similar histological features and adverse prognosis. Clin Neuropathol 37(2):53–63. https://doi.org/10.5414/NP301085
Mondal G, Lee JC, Ravindranathan A, Villanueva-Meyer JE, Tran QT, Allen SJ, Barreto J, Gupta R, Doo P, Van Ziffle J, Onodera C, Devine P, Grenert JP, Samuel D, Li R, Metrock LK, Jin LW, Antony R, Alashari M, Cheshier S, Whipple NS, Bruggers C, Raffel C, Gupta N, Kline CN, Reddy A, Banerjee A, Hall MD, Mehta MP, Khatib Z, Maher OM, Brathwaite C, Pekmezci M, Phillips JJ, Bollen AW, Tihan T, Lucas JT Jr, Broniscer A, Berger MS, Perry A, Orr BA, Solomon DA (2020) Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol 139:1071–1088. https://doi.org/10.1007/s00401-020-02155-5
Lolli V, Tampieri D, Melancon D, Delpilar Cortes M (2010) Imaging in primary central nervous system lymphoma. Neuroradiol J 23(6):680–689. https://doi.org/10.1177/197140091002300606
Guo R, Zhang X, Niu C, Xi Y, Yin H, Lin H, Chang T (2019) Primary central nervous system small lymphocytic lymphoma in the bilateral ventricles: two case reports. BMC Neurol 19(1):200. https://doi.org/10.1186/s12883-019-1430-3
Yap KK, Sutherland T, Liew E, Tartaglia CJ, Pang M, Trost N (2012) Magnetic resonance features of primary central nervous system lymphoma in the immunocompetent patient: a pictorial essay. J Med Imaging Radiat Oncol 56(2):179–186. https://doi.org/10.1111/j.1754-9485.2012.02345.x
Amrom D, Tanyalcin I, Verhelst H, Deconinck N, Brouhard GJ, Decarie JC, Vanderhasselt T, Das S, Hamdan FF, Lissens W, Michaud JL, Jansen AC (2014) Polymicrogyria with dysmorphic basal ganglia? Think tubulin! Clin Genet 85(2):178–183. https://doi.org/10.1111/cge.12141
Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, Hully M, Bianco CF, Boddaert N, Elie C, Lascelles K, Souville I, Consortium LI-T, Beldjord C, Chelly J (2014) The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 137(Pt 6):1676–1700. https://doi.org/10.1093/brain/awu082
Petrak B, Lisy J, Kraus J, Kyncl M, Zatrapa T (2008) Focal areas of high-signal intensity on brain T2-weighted magnetic resonance imaging scans are significant for the diagnosis of neurofibromatosis Von Recklinghausen type 1. Pediatrics 121(Supplement 2):S147. https://doi.org/10.1542/peds.2007-2022CCCCCC
Salman MS, Hossain S, Gorun S, Alqublan L, Bunge M, Rozovsky K (2018) Cerebellar radiological abnormalities in children with neurofibromatosis type 1: part 2 - a neuroimaging natural history study with clinical correlations. Cerebellum Ataxias 5:13. https://doi.org/10.1186/s40673-018-0092-z
Friedrich RE, Nuding MA (2016) Optic pathway glioma and cerebral focal abnormal signal intensity in patients with neurofibromatosis type 1: characteristics, treatment choices and follow-up in 134 affected individuals and a brief review of the literature. Anticancer Res 36(8):4095–4121
Manyam BV (2005) What is and what is not ‘Fahr’s disease’. Parkinsonism Relat Disord 11(2):73–80. https://doi.org/10.1016/j.parkreldis.2004.12.001
Quintans B, Oliveira J, Sobrido MJ (2018) Primary familial brain calcifications. Handb Clin Neurol 147:307–317. https://doi.org/10.1016/B978-0-444-63233-3.00020-8
Livingston JH, Stivaros S, Warren D, Crow YJ (2014) Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol 56(7):612–626. https://doi.org/10.1111/dmcn.12359
Livingston JH, Mayer J, Jenkinson E, Kasher P, Stivaros S, Berger A, Cordelli DM, Ferreira P, Jefferson R, Kutschke G, Lundberg S, Ounap K, Prabhakar P, Soh C, Stewart H, Stone J, van der Knaap MS, van Esch H, van Mol C, Wakeling E, Whitney A, Rice GI, Crow YJ (2014) Leukoencephalopathy with calcifications and cysts: a purely neurological disorder distinct from coats plus. Neuropediatrics 45(3):175–182. https://doi.org/10.1055/s-0033-1364180
Moore MM, Kanekar SG, Dhamija R (2008) Ethylene glycol toxicity: chemistry, pathogenesis, and imaging. Radiol Case Rep 3(1):122. https://doi.org/10.2484/rcr.v3i1.122
Rumboldt Z, Vavro H, Spero M (2019) Exogenuous toxins and CNS injuries. In: Barkhof F, Jäger R, Thurner M, Rovira A (eds) Clinical neuroradiology. Springer, Berlin. https://doi.org/10.1007/978-3-319-61423-6_66-1
Malhotra A, Mongelluzzo G, Wu X, Durand D, Kalra VB, LeSar B, Liu R (2017) Ethylene glycol toxicity: MRI brain findings. Clin Neuroradiol 27(1):109–113. https://doi.org/10.1007/s00062-016-0525-0
Boukobza M, Baud FJ, Gourlain H, Champion S, Malissin I, Megarbane B (2015) Neuroimaging findings and follow-up in two cases of severe ethylene glycol intoxication with full recovery. J Neurol Sci 359(1–2):343–346. https://doi.org/10.1016/j.jns.2015.11.023
Alleman AM (2014) Osmotic demyelination syndrome: central pontine myelinolysis and extrapontine myelinolysis. Semin Ultrasound CT MR 35(2):153–159. https://doi.org/10.1053/j.sult.2013.09.009
Garg P, Aggarwal A, Malhotra R, Dhall S (2019) Osmotic demyelination syndrome - evolution of extrapontine before pontine myelinolysis on magnetic resonance imaging. J Neurosci Rural Pract 10(1):126–135. https://doi.org/10.4103/jnrp.jnrp_240_18
Hussain K, Walsh TJ, Chazen JL (2016) Brain MRI findings with vigabatrin therapy: case report and literature review. Clin Imaging 40(1):180–182. https://doi.org/10.1016/j.clinimag.2015.07.016
Hussain SA, Tsao J, Li M, Schwarz MD, Zhou R, Wu JY, Salamon N, Sankar R (2017) Risk of vigabatrin-associated brain abnormalities on MRI in the treatment of infantile spasms is dose-dependent. Epilepsia 58(4):674–682. https://doi.org/10.1111/epi.13712
Perugula ML, Lippmann S (2016) Fahr’s disease or Fahr’s syndrome? Innov Clin Neurosci 13(7–8):45–46
Lee YJ, Park S, Kim YW, Park KM, Kim IH, Park JH, Park BS (2018) A case of seizure revealing Fahr’s syndrome with primary hypoparathyroidism. Am J Case Rep 19:1430–1433. https://doi.org/10.12659/AJCR.913382
Rudge P, Hyare H, Green A, Collinge J, Mead S (2018) Imaging and CSF analyses effectively distinguish CJD from its mimics. J Neurol Neurosurg Psychiatry 89(5):461–466. https://doi.org/10.1136/jnnp-2017-316853
Tschampa HJ, Zerr I, Urbach H (2007) Radiological assessment of Creutzfeldt-Jakob disease. Eur Radiol 17(5):1200–1211. https://doi.org/10.1007/s00330-006-0456-2
Maeda K, Sugihara Y, Shiraishi T, Hirai A, Satoh K (2019) Cortical hyperintensity on diffusion-weighted images as the presymptomatic marker of sporadic Creutzfeldt-Jakob disease. Intern Med 58(5):727–729. https://doi.org/10.2169/internalmedicine.1155-18
Baumann M, Hennes EM, Schanda K, Karenfort M, Kornek B, Seidl R, Diepold K, Lauffer H, Marquardt I, Strautmanis J, Syrbe S, Vieker S, Hoftberger R, Reindl M, Rostasy K (2016) Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): extending the spectrum of MOG antibody positive diseases. Mult Scler 22(14):1821–1829. https://doi.org/10.1177/1352458516631038
Pohl D, Alper G, Van Haren K, Kornberg AJ, Lucchinetti CF, Tenembaum S, Belman AL (2016) Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology 87(9 Suppl 2):S38–S45. https://doi.org/10.1212/WNL.0000000000002825
Armangue T, Olive-Cirera G, Martinez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Munoz-Batista M, Arino H, Gonzalez-Alvarez V, Felipe-Rucian A, Jesus Martinez-Gonzalez M, Cantarin-Extremera V, Concepcion Miranda-Herrero M, Monge-Galindo L, Tomas-Vila M, Miravet E, Malaga I, Arrambide G, Auger C, Tintore M, Montalban X, Vanderver A, Graus F, Saiz A, Dalmau J, Spanish Pediatric anti MOGSG (2020) Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol 19(3):234–246. https://doi.org/10.1016/S1474-4422(19)30488-0
Beattie GC, Glaser CA, Sheriff H, Messenger S, Preas CP, Shahkarami M, Venkatesan A (2013) Encephalitis with thalamic and basal ganglia abnormalities: etiologies, neuroimaging, and potential role of respiratory viruses. Clin Infect Dis 56(6):825–832. https://doi.org/10.1093/cid/cis990
Rumboldt Z (2008) Imaging of topographic viral CNS infections. Neuroimaging Clin N Am 18(1):85–92; viii. https://doi.org/10.1016/j.nic.2007.12.006
Petropoulou KA, Gordon SM, Prayson RA, Ruggierri PM (2005) West Nile virus meningoencephalitis: MR imaging findings. AJNR Am J Neuroradiol 26(8):1986–1995
Akkoc G, Kadayifci EK, Karaaslan A, Atici S, Yakut N, Ocal Demir S, Soysal A, Bakir M (2016) Epstein-Barr virus encephalitis in an immunocompetent child: a case report and management of Epstein-Barr virus encephalitis. Case Rep Infect Dis 2016:7549252–7549254. https://doi.org/10.1155/2016/7549252
Soares BP, Provenzale JM (2016) Imaging of herpesvirus infections of the CNS. Am J Roentgenol 206(1):39–48. https://doi.org/10.2214/ajr.15.15314
Smith AB, Smirniotopoulos JG, Rushing EJ (2008) From the archives of the AFIP: central nervous system infections associated with human immunodeficiency virus infection: radiologic-pathologic correlation. Radiographics 28(7):2033–2058. https://doi.org/10.1148/rg.287085135
Roche AD, Rowley D, Brett FM, Looby S (2018) Concentric and eccentric target MRI signs in a case of HIV-associated cerebral toxoplasmosis. Case Rep Neurol Med 2018:9876514–9876513. https://doi.org/10.1155/2018/9876514
Huang BY, Castillo M (2008) Hypoxic-ischemic brain injury: imaging findings from birth to adulthood. Radiographics 28(2):417–439; quiz 617. https://doi.org/10.1148/rg.282075066
Chao CP, Zaleski CG, Patton AC (2006) Neonatal hypoxic-ischemic encephalopathy: multimodality imaging findings. Radiographics 26(Suppl 1):S159–S172. https://doi.org/10.1148/rg.26si065504
Malamud MDN (1950) Status marmoratus: a form of cerebral palsy following either birth injury or inflammation of the central nervous system. J Pediatr 37(4):610–619. https://doi.org/10.1016/S0022-3476(50)80272-X
Rumboldt Z (2012) Hypertensive hematoma. In: Rumboldt Z, Castillo M, Huang BY, Rossi A (eds) Brain imaging with MRI and CT: an image pattern approach. Cambridge University Press, Cambridge, pp 364–365
Yang Z, Chen J, Mu J (2017) Simultaneous bilateral basal ganglia hemorrhage. Curr Drug Deliv 14(6):807–815. https://doi.org/10.2174/1567201813666160607202811
Yen CP, Lin CL, Kwan AL, Lieu AS, Hwang SL, Lin CN, Howng SL (2005) Simultaneous multiple hypertensive intracerebral haemorrhages. Acta Neurochir 147(4):393–399; discussion 399. https://doi.org/10.1007/s00701-004-0433-y
McKinney AM, Jagadeesan BD, Truwit CL (2013) Central-variant posterior reversible encephalopathy syndrome: brainstem or basal ganglia involvement lacking cortical or subcortical cerebral edema. AJR Am J Roentgenol 201(3):631–638. https://doi.org/10.2214/AJR.12.9677
Li K, Yang Y, Guo D, Sun D, Li C (2020) Clinical and MRI features of posterior reversible encephalopathy syndrome with atypical regions: a descriptive study with a large sample size. Front Neurol 11:194. https://doi.org/10.3389/fneur.2020.00194
Funding
No funding was received.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Ethical approval
This review article was conducted retrospectively from data obtained for clinical purposes. We consulted extensively with the IRB of Ziekenhuis Oost-Limburg who determined that our study did not need ethical approval. An IRB official waiver of ethical approval was granted from the IRB of Ziekenhuis Oost-Limburg.
Informed consent
This review article was conducted retrospectively from data obtained for clinical purposes. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
• Magnetic resonance imaging (MRI) is the modality of choice for assessment of pathology involving the deep grey matter nuclei, due to its superior contrast resolution. Nonetheless, CT has a distinct role as the reference standard for detection of calcifications.
• The basal ganglia and thalami are susceptible to injury because of the extensive metabolic activity, especially in the immature brain, due to intense neurotransmission, and therefore, pathologies often present with distinct imaging patterns, frequently in a symmetric fashion.
• A systematic approach combining imaging, clinical and demographic data is necessary when encountering lesions of the central grey matter on scans of the brain.
Rights and permissions
About this article

Cite this article
Van Cauter, S., Severino, M., Ammendola, R. et al. Bilateral lesions of the basal ganglia and thalami (central grey matter)—pictorial review. Neuroradiology 62, 1565–1605 (2020). https://doi.org/10.1007/s00234-020-02511-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-020-02511-y