
Abstract
Introduction
Embryonal tumor with abundant neuropil and true rosettes (ETANTR) is a rare subtype of primitive neuroectodermal tumors first reported in 2000. It is rare among the group of embryonal central nervous system tumors with approximately 50 reported cases. ETANTR has been suggested to be a separate entity among this group of tumors.
Case report
Herein, we present only the second autopsy case of ETANTR, which occurred in a 17-month-old boy, and was located in the brainstem. The tumor was inoperable, and despite chemotherapy, the child died 3 months after initial hospitalization. A brain only autopsy was performed.
Discussion
Neuropathological and neuroimaging examinations suggest ETANTR grew by expansion rather than invasion distorting anatomical structures of the posterior fossa. We suggest that the characteristic histopathological picture of the tumor is the result of multifocal and dispersed germinative activity surrounded by mature neuropil-like areas.
Conclusion
ETANTR is a pediatric tumor occurring in children under 4 with a significantly poor prognosis with more cases and research required to characterize this rare embryonal tumor.
Similar content being viewed by others


Introduction
Embryonal neoplasms of the central nervous system (CNS) are among the most aggressive of brain tumors in children. Embryonal tumor with abundant neuropil and true rosettes (ETANTR) is a histologically distinct embryonal tumor not yet classified as a separate entity in the 2007 World Health Organization (WHO) classification of central nervous system tumors. According to this classification, embryonal tumors are divided into three categories: medulloblastoma, atypical teratoid/rhabdoid tumor (AT/RT), and CNS primitive neuroectodermal tumors (PNET) [12, 17]. PNET is further divided into five subtypes: CNS PNET, CNS neuroblastoma, CNS ganglioneuroblastoma, medullloepithelioma, and ependymoblastoma [12, 17]. ETANTR was first described by Eberhart et al. in 2000 as a variant of pediatric embryonal brain tumors [5]. ETANTR is characterized by undifferentiated neuroepithelial cells resembling those of classic PNET, abundant well-differentiated neuropil, and ependymoblastic rosettes scattered throughout the paucicellular regions of neoplastic neuropil [7]. ETANTR is a unique subtype of CNS PNET and we would support its distinction as a separate entity within CNS PNET in future WHO classifications of embryonal CNS tumors. The number of reported cases of ETANTR remains to be low. Approximately 50 cases and less than 20 papers of ETANTR have been published. The largest single study of ETANTR was published by Gessi who reviewed 29 cases with seven original cases [7].
Herein, we present a case of ETANTR in a boy treated at the Children’s University Hospital in Cracow, Poland as well as describe previously well-known and more recently published epidemiologic, clinical, histologic, cytological, and radiologic features of ETANTR. Our case is the only the second autopsy report of ETANTR according to our review of the literature (after Kleinschmidt-DeMasters et al.), and since the integrity of the tumor was never disrupted neurosurgically, we may observe the end-stage natural history of this tumor. This tumor has a poor prognosis, and due to the paucity of cases reported so far, its true biological behavior is far from being definitely recognized. We hope that our case will increase awareness of ETANTR and could bring a contribution to the knowledge on this tumor.
Case report
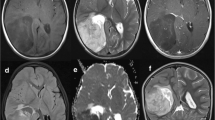
A 17-month-old boy was admitted to the Department of Neurosurgery, Children’s University Hospital in Cracow due to aggravating problems with balance. Starting a week earlier, the parents noticed that the boy had a tendency to tilt the head to the right and would choke when drinking. Neurologic examination found the following deficits: cerebellar disturbances of balance illustrated by the boy’s staggering and inability to walk, compulsory tilting of the head to the right, slight right pyramidal hemiparesis, left VI and VII cranial nerve palsies, bulbar signs demonstrated by choking, weak pharyngeal and palatal reflexes, and a suspected lesion in the visual field which could not be confirmed due to the child’s lack of cooperation. Computer tomography (CT) and magnetic resonance imaging (MRI) revealed an enormous tumor (44 × 35 × 45 mm in CT and 35 × 36 × 32 mm in MRI) in the brainstem, mostly in the left pons and medulla oblongata (Fig. 1a, b). The tumor was hyperintense in T2 and showed heterogenous contrast enhancement in T1 with narrowing of the fourth ventricle (Fig. 1b). There were no features of supratentorial hydrocephalus in spite of evidence of elevated pressure in the posterior fossa. The state of the child was unstable and the risk of its marked worsening due to operation was high. Even the biopsy bore a significant risk. As a result, the parents, after meticulous consideration of the pros and cons, did not give consent to neither an operation nor biopsy on assumption that the child would receive chemotherapy, which was immediately introduced. Consecutively, the child was transferred to the Department of Pediatric Hematology and Oncology, Children’s University Hospital in Cracow. Further diagnostics did not reveal any other foci of disease. Based on the clinical picture and neuroimaging, a malignant glioma of the brainstem was tentatively diagnosed and chemotherapy was introduced accordingly with two cycles of cisplatin, etoposide, and vincristine, and one cycle of cyclophosphamide, etoposide, and vincristine. Two months after first admission, the child showed signs of ataxia and decreased muscle tone. Soon, the child’s condition abruptly worsened with severe vomiting and a forced retroflexed position. Repeated MRI revealed enlargement of the tumor (52 × 50 × 54 mm) and supratentorial hydrocephalus due to compression of the cerebral aqueduct with transudation (Fig. 1c). The hydrocephalus was treated with implantation of a ventriculo-peritoneal shunt, and chemotherapy with irinotecan and carboplatin was introduced. The hydrocephalus remitted but the child relentlessly deteriorated as the tumor continued to enlarge and died 3 months after hospitalization.

a MRI, 3DFSPGRT1 sequence, sagittal plane. The tumor is localized in the brainstem and is extremely expanding and deforming its structure. Noteworthy is severe compression of the fourth ventricle, but evidently between the tumor and lumen of the fourth ventricle, there remains a thin, uninterrupted strip of brain tissue (arrow). b MRI, DGRET1 with contrast, axial plane. Contrast enhancement within the tumor is conspicuously insignificant. c MRI, frFSET2, coronal plane. The tumor appears as a very well delineated expansive process, with heterogeneous signal, containing some fluid-filled areas at the level of fourth ventricle and medial part of left cerebellar hemisphere, which has atrophied. The tumor molds and compresses the brainstem and obliterates the fourth ventricle. Supratentorially, there are features of hydrocephalus with transudate of CSF into surrounding brain tissue. d Macroscopic cross section of the tumor at the level of the cerebellum. Noteworthy is the very distinct border between the tumor and apparently untouched but evidently severely displaced and compressed cerebellar tissue. e Characteristic mixture of neuropil-like paucicellular zones with quite uniformly scattered cellular “densities” consisting of small undifferentiated cells. Both within densities and also directly within neuropil zones, one could discern rosettes which are quite characteristic for this tumor, H&E, objective magnification ×10. f A close-up view of rosettes. Some with empty core but delicately contoured with pinkish line, and some with homogenous featureless core (arrow), H&E, objective magnification ×40
An autopsy of the brain performed at Department of Pathology, Children’s University Hospital, revealed an enormous tumor inside left cerebellar hemisphere partially occupying the pons and also expanding to the midbrain. The tumor distorted the whole brainstem and cerebellum, pushing aside and compressing the fourth ventricle, but definitely not originating from it (Fig. 1d). This confirmed the previous MRI findings of a primary brainstem tumor (Fig. 1a). On cross section, the tumor showed uniform consistency, slightly lower than the normal brain tissue. The tumor had well marked borders with a smooth transition into adjacent apparently normal tissue.
Microscopic pictures represented a very conspicuous pattern with relatively paucicellular neuropil-like background with numerous and rather evenly distributed cellular densities populated with small undifferentiated cells (Fig. 1e). Rosettes formed another characteristic element of the tumor (Fig. 1f). Neuropil zones, in contrast to the cellular zones, showed immunopositivity to synaptophysin (Fig. 2a). Both in cellular zones and neuropil zones, there were scattered characteristic rosettes, totally negative to synaptophysin (Fig. 2b) but strongly positive to vimentin (Fig. 2c). The rosettes had an “empty,” homogenous core, but frequently with a delicate eosinophilic contouring reminiscent of ependymoblastic rosettes, though somewhat more delicate than those occurring in typical ependymoblastoma (Fig. 1e, f). The rosettes were also weakly positive for glial fibrillary acidic protein (GFAP) and for S100 protein (Fig. 2d). However, in neuropil zones, there were cells present with the immuno- and morphophenotype of astrocytes (GFAP+ and some S100+). Moreover, outside of the rosettes, synaptophysin-positive mature neuron-like cells were noted (Fig. 2b), and some were even strongly positive for synaptophysin, in spite of resembling astrocytes (Fig. 2e). Ki-67 expression was high but limited to rosettes (Fig. 2f) and cellular zones especially around the vessels. No areas of frank necrosis or any “pathological” forms of endothelial proliferation were noted. However delicate vessels were numerous, frequently surrounded by undifferentiated cells that do not conform to the description of “perivascular formations” or pseudorosettes.

a The more cellular zones (asterisk), in contrary to neuropil background, are rather negative for synaptophysin, objective magnification ×20. b All rosettes were negative for synaptophysin. Apart from the rosettes, strongly synaptophysin-positive cells with prominent processes resembling astrocytes rather than neurons were noted, objective magnification x40. c Both rosettes and almost all cells within cellular “densities” are strongly immunopositive for vimentin, objective magnification ×40. d Rosettes are negative for S100. However, between the rosettes there are S-100 positive cells with marked processes, objective magnification ×20. e Some mature looking synaptophysin-positive neurons (arrowheads) are scattered within neuropil, objective magnification ×40. f Proliferating activity as evidenced by immunopositivity to Ki67 was marked within rosettes and also around vessels (not shown in this picture), objective magnification ×40
Discussion
The neuropathological findings in the presented case of ETANTR, especially the very distinct border between tumor and apparently normal tissue, could be observed in the whole cross section of the brain during autopsy examination. This suggests that this tumor rather expands within the cerebral/cerebellar tissue rather than invades it. If so, most if not all cellular elements within tumor are of neoplastic origin (probably including a de novo formed vascular bed). One may speculate that cellular “densities” and in particular rosettes supposedly recapitulating primitive neural tube represent germinative centers, and in turn, the neuropil paucicellular zones are essentially the result of maturation. The maturation is evidently multidirectional as for the cell lineages since within the tumor one can find ganglion cells as well as glia, and probably also “hybrid” cells like the ones that show strong synaptophysin immunoreactivity and astrocytic-like morphology (Fig. 2b). As a result, ETANTR can justifiably be recognized as an embryonal tumor with a characteristic multifocal germinative process. In spite of the remarkable size of tumor, there was no evidence of a spread, neither hematogenous nor via CSF. ETANTR has exhibited metastases within the neuroaxis in only a minority of cases [7].
ETANTR was first described by Eberhart et al. in 2000 as a variant of pediatric embryonal brain tumors [5]. ETANTR occurs in children aged 4 and under, mostly in children under 2, and is more common in girls, unlike the other CNS embryonal tumors, which occur mostly or equally in boys [7]. Most are located in the supratentorial region, occasionally infratentorially, and rarely in the spinal cord [7]. In addition to our case report, clinical symptoms and features of ETANTR include increased intracranial pressure, seizures, hemiparesis, cerebellar signs, cranial nerve palsies, and other neurologic deficits [14].
Microscopically, ETANTR is characterized by undifferentiated neuroepithelial cells resembling those of classic PNET, abundant well-differentiated neuropil, and ependymoblastic rosettes scattered throughout paucicellular regions of neoplastic neuropil [7]. Homer Wright, Flexner–Wintersteiner, and pseudovascular rosettes may also appear. Unlike AT/RT, ETANTR has no rhabdoid cells, and unlike medulloepithelioma, there are no epithelial-like formations [14]. ETANTR has distinct ependymoblastic rosettes in both hypercellular and acellular regions. The hypercellular regions have small blue hyperchromic cells with a round nuclei and indistinct cell borders [7]. Al-Hussain described rhabdomyoblastic and melanocytic differentiation in a single case of ETANTR which had not been reported before [1].
Numerous cytogenetic studies have been performed on ETANTR in an effort to uniquely identify this tumor. These include polysomy of chromosome 2 [3, 7, 16] and abnormalities in chromosome 17 including isochromosome 17q and polysomy 17 [6]. Such abnormalities of chromosome 17 are also found in medulloblastoma [13], while polysomy of chromosome 2 appears to be a unique differentiating feature of ETANTR [2]. Amplification of 19q13.42 with upregulation of microRNA clusters and protein-coding genes has also been noted as a unique feature [16]. Korshunov et al. identified 19q13.42 amplification in 95 % of ETANTR and 90 % of ependymoblastomas [10], and more recently, Nobusawa et al. identified 19q13.42 amplification in three out of four ETANTR, one medulloepithelioma with ETANTR components, and one ependymoblastoma [15].
In MRI, ETANTR presents as a well-demarcated solid mass with surrounding edema often producing marked mass effects. Most tumors are solid ranging from 2 to 8 cm in diameter, while few may show a cystic component and microcalcifications [7, 14]. The tumor appears hypointense on T1 and hyperintense on T2 with varying enhancement as cases of homogenously, heterogeneously, and non-enhancing lesions have been reported [7, 14]. MR spectroscopy reveals a choline peak and a high ratio of choline/aspartate suggesting hypercellularity of the tumor [7]. The tumor grossly appears as a pinkish white neoplasm infiltrating the surrounding brain parenchyma with occasional well-demarcated borders. Five cases with dural attachment have been reported [7, 14]. Spread occurs through the cerebrospinal fluid often with leptomeningeal metastases [9, 14]. Most tumors were resectable upon first presentation although recurrences were common. Current treatment strategy revolves around complete tumor resection, systemic chemotherapy, and craniospinal radiation when appropriate. Overall, few children remain free of disease such as the 4 of 29 cases reviewed by Gessi [7]. Most recent studies show that 76 % of patients have died with a median survival of 9 months [7]. Hope for tumor-free survival is evident though as a case presented by Manjila remains alive with no signs of recurrences 7 years after surgery [13].
ETANTR shares numerous features of ependymoblastoma and even some authors have suggested removing the diagnosis of ependymoblastoma from the WHO classification of CNS tumors due to its lack of specificity and claims of obsolescence. Judkins found eight instances of ependymoblastoma that upon further review with proper sampling were rediagnosed as ETANTR [8]. Korshunov revealed high cytogenetic homology between ETANTR and ependymoblastoma [10].
As mentioned, the number of reported cases of ETANTR is low. Approximately 50 cases and less than 20 papers of ETANTR have been published. The largest single study of ETANTR was published by Gessi who reviewed 29 cases with seven original cases [7]. Cases reviewed by Gessi and additional cases were reported by Niguez [14], Manjila [13], Buccoliero [3], Pfister [16], Al-Hussain and Dababo [1], Dunham [4], Fuller [6], La Spina [11], and other authors. Only one autopsy case apart from our own has been published [9]. Prognosis of ETANTR is poor which is exemplified by the 21 deaths of 29 cases reported by Gessi [7], death of both cases reported by Niguez [14], death of one of two cases reported by Manjila [13], and the fatal case presented here.
Conclusion
Our case together with other cases of ETANTR reveals that ETANTR has a unique and specific histopathologic and cytologic profile that may be recognized as a unique entity within CNS PNET. It is characterized by undifferentiated neuroepithelial cells, abundant well-differentiated neuropil, and ependymoblastic rosettes abruptly arising from paucicellular regions of neoplastic neuropil. Our case of ETANTR investigated by autopsy suggests that the tumor may develop for a substantial time by expansion rather than true invasion. In our opinion, ETANTR appears to be a tumor with multifocal germinative activity and maturation leading to its signature appearance of hypercellular rosettes dispersed within the neuropil. Prognosis is poor with approximately 76 % of patients succumbing to disease and only five patients have no evidence of disease. In the 12 years since its first description in 2000, approximately 50 cases and less than 20 papers on ETANTR are present in the literature. A call for more cases of ETANTR is made by us and many other authors in order to better characterize this often fatal tumor’s behavior and response to both surgical and chemotherapeutic treatment protocols.
References
Al-Hussain TO, Dababo MA (2009) Posterior fossa tumor in a 2-year-old girl. Brain Pathol 19:343–346
Biegel JA, Allen CS, Kawasaki K, Shimizu N, Budarf ML, Bell CJ (1996) Narrowing the critical region for a rhabdoid tumor locus in 22q11. Genes Chromosomes Cancer 16:94–105
Buccoliero AM, Castiglione F, Rossi Degl'Innocenti D, Franchi A, Paglierani M, Sanzo M, Cetica V, Giunti L, Sardi I, Genitori L, Taddei GL (2010) Embryonal tumor with abundant neuropil and true rosettes: morphological, immunohistochemical, ultrastructural and molecular study of a case showing features of medulloepithelioma and areas of mesenchymal and epithelial differentiation (case report). Neuropathology 30:84–91
Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocystic differentiation. J Neurooncol 84:91–98
Eberhart CG, Brat DJ, Cohen KJ, Burger PC (2000) Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 3:346–352
Fuller C, Fouladi M, Gajjar A, Dalton J, Sanford RA, Helton KJ (2006) Chromosome 17 abnormalities in pediatric neuroblastic tumor with abundant neuropil and true rosettes. Am J Clin Pathol 126:277–283
Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W, Hawkins C, Rosenblum MK, Burger PC, Eberhart CG (2009) Embryonal tumors with abundant neuropil and true rosettes. A distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33:211–217
Judkins AR, Ellison DW (2008) Ependymoblastoma: dear, damned, distracting diagnosis, farewell! Brain Pathol 20:133–139
Kleinschmidt-DeMasters BK, Boylan A, Capocelli K, Boyer PJ, Foreman NK (2011) Multinodular leptomeningeal metastases from ETANTR contain both small blue cell and maturing neuropil elements. Acta Neuropathol 122(2011):783–785
Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H, Tobias V, Buccoliero AM, Sardi I, Gardiman MP, Bonnin J, Scheithauer B, Kulozik AE, Witt O, Mork S, von Deimling A, Wiestler OD, Giangaspero F, Rosenblum M, Pietsch T, Lichter P, Pfister SM (2010) Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120:253–260
La Spina M, Pizzolitto S, Skrap M, Nocerino A, Russo G, Di Cataldo A, Perilongo G (2006) Embryonal tumour with abundant neuropil and true rosettes. A new entity or only variations of a parent neoplasm (PNETs)? This is a dilemma. J Neurooncol 78:317–320
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) WHO classification of tumors of the central nervous system. Acta Neuropathol 114:97–109
Manjila S, Ray A, Hu Y, Cai DX, Cohen ML, Cohen AR (2011) Embryonal tumors with abundance neuropil and true rosettes: 2 illustrative cases and a review of the literature. Neurosurg Focus 30(1):E2
Niguez BF, Martínez-Lage JF, Almagro MJ, Fuster JL, Serrano C, Torroba MA, Sola J (2010) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): a new distinctive variety of pediatric PNET: a case-based update. Childs Nerv Syst 26:1003–1008
Nobusawa S, Yokoo H, Hirato J, Kakita A, Takahashi H, Sugino T, Tasaki K, Itoh H, Hatori T, Shimoyama Y, Nakazawa A, Nishizawa S, Kishimoto H, Matsuoka K, Nakayama M, Okura N, Nakazato Y (2012) Analysis of chromosome 19q13.42 amplification in embryonal brain tumors with ependymoblastic multilayered rosettes. Brain Pathol 22:689–697
Pfister S, Remke M, Castoldi M, Bai AH, Muckenthaler MU, Kulozik A, von Deimling A, Pscherer A, Lichter P, Korshunov A (2009) Novel genomic amplification targeting microRNA cluster 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol 117:457–464
WHO (2007) Classification of tumors of the central nervous system, 4th edn. IARC, Lyon, pp 8–9
Conflict of interest
The authors report that informed parental consent has been obtained for autopsy and inclusion in this case report. The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Adamek, D., Sofowora, K.D., Cwiklinska, M. et al. Embryonal tumor with abundant neuropil and true rosettes: an autopsy case-based update and review of the literature. Childs Nerv Syst 29, 849–854 (2013). https://doi.org/10.1007/s00381-013-2037-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-013-2037-4