
Abstract
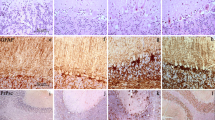
We studied the immunocytochemical distribution of the prion or proteinase-resistant protein (PrP) during the evolution of experimental Creutzfeldt-Jakob disease (CJD) in mice. Fifty-one brains were collected up to 22 weeks following intracerebral inoculation with the Fujisaki strain of the CJD agent. Slides were also immunostained for apolipoprotein E (apoE) and glial fibrillary acidic protein. Vacuolar changes with focal astrocytosis first occurred around the needle track at week 2 and later spread along white matter tracks. Until week 12, changes were asymmetrical, affecting more the side of inoculation. Spongiform change and astrogliosis spread subsequently to the gray matter. Time course and intensity of spongiform change and immunocytochemistry for PrP were discrepant: in most brain regions, severe vacuolation preceded immunocytochemically detectable PrP accumulation. PrP deposits in form of small dots were first detectable at week 6 in the area surrounding the needle track. After week 7, plaque-like amorphous PrP deposits were observed in white matter pathways. Finally, PrP was detectable also in basal ganglia and in the dorsal hippocampus (week 13) and in the neocortex (week 17), as the synaptic type of PrP immunopositivity. In the hippocampus, diffuse PrP deposits paralleled spongiform change, while in the cortex severe vacuolation was accompanied only by weak synaptic PrP deposits. Immunocytochemically detectable apoE was restricted to compact plaque-type PrP deposits after week 15. We conclude that disease-specific neuropathology spreads from the needle track along white matter pathways towards the gray matter; in this model, there is some discrepancy between development of tissue pathology and immunocytochemically detectable deposition of PrP. Immunocytochemically detectable apoE deposition follows PrP accumulation.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 22 December 1998 / Revised, accepted: 6 April 1999
Rights and permissions
About this article
Cite this article
Kordek, R., Hainfellner, J., Liberski, P. et al. Deposition of the prion protein (PrP) during the evolution of experimental Creutzfeldt-Jakob disease. Acta Neuropathol 98, 597–602 (1999). https://doi.org/10.1007/s004010051124
Issue Date:
DOI: https://doi.org/10.1007/s004010051124