
Abstract
The objective of this study is to evaluate the effectiveness and safety of total disc replacement surgery compared with spinal fusion in patients with symptomatic lumbar disc degeneration. Low back pain (LBP), a major health problem in Western countries, can be caused by a variety of pathologies, one of which is degenerative disc disease (DDD). When conservative treatment fails, surgery might be considered. For a long time, lumbar fusion has been the “gold standard” of surgical treatment for DDD. Total disc replacement (TDR) has increased in popularity as an alternative for lumbar fusion. A comprehensive systematic literature search was performed up to October 2008. Two reviewers independently checked all retrieved titles and abstracts, and relevant full text articles for inclusion. Two reviewers independently assessed the risk of bias of included studies and extracted relevant data and outcomes. Three randomized controlled trials and 16 prospective cohort studies were identified. In all three trials, the total disc replacement was compared with lumbar fusion techniques. The Charité trial (designed as a non-inferiority trail) was considered to have a low risk of bias for the 2-year follow up, but a high risk of bias for the 5-year follow up. The Charité artificial disc was non-inferior to the BAK® Interbody Fusion System on a composite outcome of “clinical success” (57.1 vs. 46.5%, for the 2-year follow up; 57.8 vs. 51.2% for the 5-year follow up). There were no statistically significant differences in mean pain and physical function scores. The Prodisc artificial disc (also designed as a non-inferiority trail) was found to be statistically significant more effective when compared with the lumbar circumferential fusion on the composite outcome of “clinical success” (53.4 vs. 40.8%), but the risk of bias of this study was high. Moreover, there were no statistically significant differences in mean pain and physical function scores. The Flexicore trial, with a high risk of bias, found no clinical relevant differences on pain and physical function when compared with circumferential spinal fusion at 2-year follow up. Because these are preliminary results, in addition to the high risk of bias, no conclusions can be drawn based on this study. In general, these results suggest that no clinical relevant differences between the total disc replacement and fusion techniques. The overall success rates in both treatment groups were small. Complications related to the surgical approach ranged from 2.1 to 18.7%, prosthesis related complications from 2.0 to 39.3%, treatment related complications from 1.9 to 62.0% and general complications from 1.0 to 14.0%. Reoperation at the index level was reported in 1.0 to 28.6% of the patients. In the three trials published, overall complication rates ranged from 7.3 to 29.1% in the TDR group and from 6.3 to 50.2% in the fusion group. The overall reoperation rate at index-level ranged from 3.7 to 11.4% in the TDR group and from 5.4 to 26.1% in the fusion group. In conclusion, there is low quality evidence that the Charité is non-inferior to the BAK cage at the 2-year follow up on the primary outcome measures. For the 5-year follow up, the same conclusion is supported only by very low quality evidence. For the ProDisc, there is very low quality evidence for contradictory results on the primary outcome measures when compared with anterior lumbar circumferential fusion. High quality randomized controlled trials with relevant control group and long-term follow-up is needed to evaluate the effectiveness and safety of TDR.
Similar content being viewed by others

Introduction
Low back pain is a major health problem in Western countries [1, 2]. A variety of pathologies can cause low back pain, one of which is degenerative disc disease (DDD) [3]. It has been hypothesised that through disc dehydration, annular tears, and loss of disc height or collapse, DDD can result in abnormal motion of the segment and biomechanical instability causing pain [4–7].
When conservative treatment fails, patients and health care providers may consider other treatment options such as surgery. Although the rationale for surgery is often not clear and despite the lack of convincing evidence in the literature regarding the effectiveness of surgery in the treatment of symptomatic DDD, the number of surgical procedures performed is continually increasing [8, 9]. For a long time, lumbar fusion (arthrodesis) has been the “gold standard” surgical treatment for DDD. However, long-term results are poor and complications common [4, 10].
An alternative surgical procedure, total disc replacement, has increased in popularity. The purpose of this technique is to restore and maintain spinal segment motion, which is assumed to prevent adjacent level degeneration at the operated levels, while relieving pain [4, 11–13].
Replacing a degenerated joint instead of fusing it was considered for the spine due to the success of total knee and hip arthroplasty [5, 14, 15]. The first described total disc replacement was the Fernstorm steelball endprosthesis in the late 1950s [16]. Since that time, multiple disc replacement prostheses have been designed for use in the lumbar spine. A large majority would never be implanted in humans [4, 10, 17]. The first prosthesis designed to be commercially distributed as an artificial disc was initiated in 1982 by Schellnack and Buttner-Janz. Currently, many different lumbar total disc prostheses are available and approved for the European market. In the United States, American Investigational Device Exemption (IDE) trials have let to FDA approval for Charité and Prodisc prostheses.
In this article, we systematically review the available literature on the effectiveness and safety of currently available prostheses for TDR in patients with systematic DDD.
Materials and methods
Objective
The objective of this systematic review was to assess the effectiveness and safety of total disc replacement surgery in patients with chronic low back pain due to DDD. The main research questions were:
-
1.
What is the course of DDD complaints and/or symptoms following total disc replacement surgery?
-
2.
What is the effectiveness of total disc replacement surgery compared to other treatments?
-
3.
What is the safety of total disc replacement surgery?
For this systematic review, we used the method guidelines for systematic reviews as recommended by the Cochrane Back Review Group [18]. Below the search strategy, selection of the studies, data extraction, risk of bias assessment, and data analysis are described in more detail. All these steps were performed by two reviewers independently (KvdE and RO) and during consensus meetings, potential disagreements between the two reviewers regarding these issues were discussed. If they were not resolved a third reviewer (MvT) was consulted.
Search strategy
An experienced librarian performed a comprehensive systematic literature search. The MEDLINE, EMBASE and COCHRANE LIBRARY databases were searched for relevant studies from 1973 to October 2008. The search strategy consisted of a combination of keywords concerning the technical procedure (e.g. disc replacement, prosthesis, implantation, discectomy, arthroplasty) and keywords regarding the anatomical features and pathology (e.g. intervertebral disc degeneration, discitis, low back pain, lumbosacral region, lumbar vertebrae). These keywords were used as MESH headings and free text words. In addition, a search was performed using the specific names of the prostheses. The full search strategy is available upon request.
Selection of studies
The search was limited to studies published in English, German, and Dutch, because these are the languages that the review authors are able to read and to understand. Two review authors independently examined all titles and abstracts that met our search terms and reviewed full publications, when necessary. The reference section of all primary studies was inspected for additional references. For the assessment of the course of complaints and/or symptoms (research question 1), we included prospective cohort studies reporting on at least 20 cases and having a follow up period of more than 6 weeks. By definition, cohort studies do not provide information about effectiveness, so for assessment of the effectiveness (research question 2), we only included randomized controlled trials. When multiple articles were identified on the same study, but describing different follow up measurements, they were included. However, articles describing only one arm of the trial, or only describing the results of 1 centre of a multicentre trial were excluded. For assessing the safety (research question 3), we extracted data on all reported complications from the prospective cohort studies and randomized controlled trials we included for research questions 1 and 2. Furthermore, we included overview studies on complications. Case reports were excluded.
Data extraction
Two review authors independently extracted relevant data from the included studies regarding design, population (e.g. age, gender, duration of complaints), type of total disc replacement surgery, type of control intervention (e.g. no treatment, lumbar fusion), vertebral level(s) operated on, follow-up period, and outcomes. Primary outcomes that were considered relevant were pain intensity [e.g. visual analogue scale (VAS), functional status, e.g. Roland Morris Disability Scale, Oswestry Scale (ODI), global improvement and return to work]. All ODI scores and VAS scores were converted into 0–100 scale. Other outcome measures, such as physiological outcome, radiological outcomes, and patient satisfaction were considered as secondary outcome measures.
Risk of bias (RoB) assessment
Two review authors independently assessed the risk of bias (RoB) of the included studies. Controlled trials were assessed using a criteria list recommended by the Cochrane Back Review Group [18]. The following criteria are scored yes, no or unsure: (1) Was the method of randomization adequate? (2) Was the treatment allocation concealed? (3) Was the patient blinded to the intervention? (4) Was the care provider blinded to the intervention? (5) Was the outcome assessor blinded to the intervention? (6) Was the dropout rate described and acceptable? (7) Were all randomized participants analyses in the group to which they were allocated? (8) Are reports of the study free of suggestion of selective outcome reporting? (9) Were the groups similar at baseline regarding the most important prognostic indicators? (10) Were co-interventions avoided or similar? (11) Was compliance acceptable in all groups? (12) Was the timing of the outcome assessment similar in all groups? Criteria 11 was scored not applicable because we consider compliance not relevant for surgical interventions. If studies met at least 6 of the 12 items, the RoB was considered low. Disagreements were resolved in a consensus meeting and a third review author was consulted when necessary. Full assessment is available upon request. The overall grading of the evidence was based on the GRADE approach [19].
Results
Search and selection
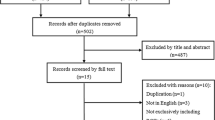
A total of 1,962 references were identified from MEDLINE, EMBASE and the COCHRANE LIBRARY that were potentially relevant for this review on total disc replacement surgery. After checking titles and abstracts, a total of 112 full text articles were retrieved that were potentially eligible for answering all research questions. After reading full text, 21 articles reporting on 16 studies were relevant for answering research question 1, and 16 articles reporting on 3 studies were relevant for research question 2. Seven overview articles for answering research question 3. Figure 1 shows the search strategy process in a flow diagram. Reviewing the reference lists of these articles resulted in no additional studies.

Flow diagram
Type of studies
For assessing the course of DDD complaints and/or symptoms (research question 1) 16 prospective cohort studies were included, describing four different devices, 6 for Charité [20–26], 8 for Prodisc [27–36], 1 for Maverick [37–39], and 1 for Acroflex [40]. For assessing the effectiveness of total disc replacement (research question 2), we identified three randomized controlled trials, all conducted in the USA in order to get FDA approval. Each trial examined a different prostheses for TDR (Charité, and ProDisc and Flexicore) and all used fusion (although different types) as control intervention. No studies comparing TDR surgery to other treatments were found. Two trials (Charité and ProDisc trial) were described in multiple articles [6, 10, 13, 41–53]. Given our inclusion and exclusion criteria, we finally included two articles describing the Charité trial; one reporting the 24 months follow up [10] and the other the 5-year follow up [51]. However, 5-year follow-up results were only available of 57% of the originally randomized population. We included one article for the ProDisc trial reporting the 24-month follow-up results [52]. The Flexicore trial was described in one article, which should be considered as preliminary results as the final results of this trial have not yet been published [53] (Table 1).
Assessment of risk of bias
After assessing, the risk of bias of the controlled trials there was 20% disagreement between the two review authors. Consultation of the third reviewer was not necessary because disagreements were resolved in a consensus meeting. For the 24 months follow up, the reporting on the Charité trial was considered to have a low risk of bias. However, the reporting on the 5-year follow up was considered to have a high risk of bias. The reporting on the ProDisc trial was considered to have a high risk of bias as it only met 4 out of the 11 risk of bias criteria. The Flexicore study was also considered to have a high risk of bias as it only met 2 out of 11 risk of bias criteria (Table 2). By design, the prospective cohort studies were not only included in the effectiveness analysis, but also used to describe the course of DDD complaints and/or symptoms after undergoing a TDR surgery.
Outcomes
(1) What is the course of DDD complaints and/or symptoms following total disc replacement surgery?
Charité® (Table 3)
The Charité prostheses is the first total disc prostheses, developed by Butter-Janz and Schellnack at the Charité clinic in former East Germany. The CharitéIII became commercially available for the first time in the late 1980s [54, 55]. Lemaire et al. reported 2 articles, respectively, with 51 months follow-up in 1997 [20] and with 11.3 years follow up in 2005 [23] on the same population. These articles report a good or excellent clinical result, respectively, in 85 and 90%. Several other prospective cohort studies report positive results as well on VAS improvement (range 16–66 points), ODI improvement (range 14–51%) and patients’ satisfaction (range 69–92%) [21–26].
ProDisc® (Table 3)
The ProDiscI was developed in France by Marnay, who operated on 64 patients and performed a single or multi-level total disc replacement in the beginning of the 1990s [29, 54, 55]. Fifty-five patients were available for follow-up after average 8.7 years. 82.6% of the patients were “completely satisfied” or “satisfied” with the results [29]. ProDiscII, the second generation ProDisc, is reported on in several publications with follow up range 3 months to 2 years [27, 28, 30–36]. Primary outcome results suggest being positive, VAS improvement (range 40–62 points) and ODI improvement (range 21–48%). Moreover, the majority of patients seem to be satisfied with the results (range 79–100%).
Maverick® (Table 3)
Huec et al. published several studies on the Maverick device [37–39]. At 2-year follow-up improvement was reported in VAS for low back pain and leg pain, decreasing by 44 and 18 points, respectively. Functional status improved as ODI score decreased 20.7%, and an overall improvement in functional status of 25% occurred in 75% of the patients. Since 2003, a prospective controlled trial has been ongoing in the USA [54].
Acroflex® (Table 3)
Fraser et al. [40] conducted two pilot studies and combined the results. The endplates were changed for the second study because of device failure. For the whole group, the functional impairment (ODI) improved from 14.9% and the Low Back Pain Score (LBPS) improved from 17.7 to 33 at 24 months follow-up. 50% of the patients were not working because of their back condition. Due to detection of mechanical failure, the randomized controlled trial has not been carried out.
In conclusion, many studies suggest pain relief, improvement in functional status and patient satisfaction after TDR surgery. The overall outcome is positive, reduction of pain intensity (range 16–66 points) and improvement of functional impairment (range 14–51%). Moreover, the majority of patients seem to be satisfied with the results (range 69–100%). Unfortunately, detailed information on how outcomes were measured was often lacking. Although outcome results from observational studies suggest a positive course after TDR surgery, a drawback is that a significant amount of complications was reported as well (which will be described later), and a control group was lacking in these studies.
(2) What is the effectiveness of total disc replacement surgery compared to other treatments?
Charité® trial (Table 1)
The Charité trial [10, 51], which was designed as a non-inferiority trail, randomized 304 patients to either TDR with the Charité III disc (n = 205) or anterior interbody fusion with BAK cage (n = 99) with a follow-up of 2 and 5 years. The primary outcomes were pain (VAS), functional impairment (ODI), overall clinical success (defined by using four criteria: ≥25% improvement in ODI, device failure, major complications, and neurological deterioration). As a secondary outcome, patient satisfaction was measured. The improvements on pain intensity (−40.6 vs. −34.1) and functional impairment (24.3 vs. 21.6%), for the TDR and the BAK, respectively, did not differ significantly at 2-year follow up. The overall clinical success (indeed statistically tested on non-inferiority) revealed that the Charité group was non-inferior to the lumbar fusion group (57.1 vs. 46.5%; P < 0.0001. P value based on the Blackwelder’s test for equivalence). Patient satisfaction was significantly better in the Charité group (73.7%) compared to the control group (53.1%) (P < 0.002). 5-year results, based on only 57% of the randomized patients and with a high risk of bias, were broadly in line with the 2-year results. At 5-year follow-up, outcomes on the composite score of clinical success showed that the Charité was non-inferior to the lumbar fusion group (57.8 vs. 51.2%; P < 0.04. P value based on the Blackwelder’s test for equivalence) [51]. There were no statistically significant differences in functional impairment and pain intensity. In conclusion, there is low quality evidence (based on one study only with a low risk of bias) that there are no clinically relevant differences on the primary outcome measures between the Charité group and the BAK cage at the 2-year follow up, and there is very low quality evidence (based on 1 study only with a high risk of bias) that there are no clinical relevant differences on the primary outcome measures at the 5-year follow up.
ProDisc® trial (Table 1)
The ProDisc trial [52], which had a high risk of bias, randomized 236 patients to either TDR with the ProDisc device (n = 161) or to anterior lumbar circumferential fusion (using femoral ring allograft and posterolateral fusion with autogenous iliac crest bone graft in combination with pedicle screws) (n = 75). Outcomes were reported with 2-year follow-up. Clinical success was defined using a combination of 10 outcomes as required by the FDA (Oswestry ≥ 15 points, SF-36 improvement, device success, neurologic success and six radiographic outcomes: no migration, no subsidence, no radiolucency, no loss of disc height, fusion status and ROM). Clinical success was statistically significantly better in the ProDisc (54.3%) than the fusion group (40.8%) (P < 0.05). Although this trail was designed as a non-inferiority study, it is unclear what statistical testing is applied. However, there were no significant differences between both groups on the mean functional impairment (−28.9 vs. −22.9%) and pain intensity scores (−39 vs. −32). In conclusion, there is very low quality evidence (based on 1 study only with a high risk of bias and inconsistent findings) for contradictory results on the primary outcome measures at the 2-year follow up for the ProDisc when compared with anterior lumbar circumferential fusion.
Flexicore® trial (Table 1)
The Flexicore trial [53], with a high risk of bias reported the initial results of 76 patients from two clinics involved in a randomized multicentre controlled trial comparing the Flexicore device (n = 44) versus anterior lumbar circumferential fusion (n = 23) with 2 year follow-up. These 76 patients are only a small proportion of all randomized patients (n = 401) included in the complete trail. Overall, dropout rate was high, 33 patients (75%) in the intervention group and 16 patients (70%) in the control group after two years. Improvement in pain intensity (VAS −70 vs. −62) and functional impairment (Oswestry −56 vs. −46%) was slightly better in the Flexicore group than in the fusion group, but the authors did not report whether this difference was statistically significant or not. Because these are preliminary results, in addition to the high risk of bias, we refrain from drawing conclusions based on this study. In general, these results suggest no clinical relevant differences between TDR surgery and fusion techniques and a small overall success rate in both groups (approximately 50%).
(3) What is the safety of total disc replacement surgery?
Although some studies reported no major complications, other cohort studies describe a wide range (1.0–91.0%) of complication rates following TDR. The majority of these studies reported complication rates ranging from 10 to 40%. Complications can be separated into those related to the surgical approach (e.g. vascular injury, nerve root damage, retrograde ejaculation) range from 2.1 to 18.7%, related to the prosthesis (e.g. subsidence, migration, implant displacement, implant failure, end plate fracture) range from 2.0 to 39.3% and related to the treatment (e.g. wound, pain, neuromusculoskeletal) range from 1.9 to 62.0%. General surgical related complications ranged from 1.0 to 14.0%. Reoperation at index level was seen in 1.0–28.6% (Table 4). These reported complication rates and reoperation rates have to be interpreted carefully, because they have been described poorly.
Below we will describe the complications rates and re-operation rates as found in the three trials. The Flexicore trial [53] report 22.7% complications in the TDR group and 43.5% in the fusion group. Reoperations are reported in both groups; 11.4% for TDR and 26.1% for fusion (Table 5).
In the Charité trial, overall complication rates published by Blumenthal et al. [10] were 29.1% for TDR and 50.2% for fusion at 2-year follow-up. Device failures necessitating reoperation were reported in 5.4% of patients in de TDR group and 9.1% of patients in the fusion group at 2 year follow-up (Table 5). However, in the FDA report on the Charité trial much higher scores of adverse effects (TDR group 181.9% and fusion group 189.6%) were reported [56]. In an article from McAfee et al. [57], analysing the incidence of reoperations, even higher reoperation rates in the Charité trial are reported (6.3% in de TDR group and 10.1% in the fusion group).
In the ProDisc trial, there was a similar discrepancy between the article and the FDA report. The overall complication rate as reported by Zigler et al. [52] were 7.3% and 6.3% for TDR and fusion, respectively, but in the FDA report on the ProDisc trial much higher scores on adverse events were reported (TDR group 255.5% and fusion group 270.7%) [58]. Reoperation was necessary for 3.7% TDR patients and 5.4% fusion patients according to Zigler et al. The number of patients needed a reoperation was similar in the FDA report; however, the included number of patients in the trial was higher so the percentage of reoperation in the FDA rapport was slightly higher (Table 5).
Geisler et al. [59] analysed only the neurological complications in the Charité trial. The incidence was no higher in patients with the Charité (16.6%) than patients with BAK fusion (17.2%) (P > 0.3). Major neurologic complications in the Charité group (e.g. burning or dysesthetic leg pain, motor deficit in index level, nerve root injury) were reported in 4.9% and in the fusion group (e.g. burning or dysesthetic leg pain, motor deficit at the index level) in 4%. One device related major complication, nerve root injury, was reported in the TDR group.
Leary et al. [60] reported on 18 patients requiring an anterior revision procedure for repositioning or removal of the Charité prosthesis because of complications. Three patients required revision of two levels. One patient had both levels revised in a single procedure, whereas two patients required staged procedures in order to revise both implants. Therefore, 21 implants were revised via 20 anterior procedures in 18 patients. Six revision cases were performed within the early postoperative period (7–14 days), all as a result of implant migration or dislocation. Late revision cases were required in 14 cases (range 3 weeks–4 years) due to implant migration, dislocation, end plate fractures, subsidence or persistent low back pain.
Van Ooij et al. [61–65] reported in several publications patients following implantation of the Charité prosthesis who experienced complications. Over the last 10 years, 75 patients with persisting back and leg pain and being unsatisfied with their clinical condition have been seen and analysed. An overview on late complications after TDR: subsidence (n = 39), prosthesis too small (n = 24), adjacent disc degeneration (n = 36), degenerative scoliosis (n = 11), facet joint degeneration on CT scan (n = 25), anterior migration (n = 6), posterior migration (n = 2), breakage metal wire (n = 10), wear (n = 5), severe osteolysis (n = 1), subluxation PE core (n = 1). 46 out of these 75 needed one or more salvage operations after their TDR. Fifteen patients were receiving posterior fusion without removal of the prosthesis. Because of persisting pain, afterwards 4 patients had their prosthesis removed in an additional operation. In 22 patients, 26 prostheses were removed and an anterior and posterior fusion was performed. In addition, seven patients received posterior fusion elsewhere, and in two patients, the disc prosthesis was removed elsewhere. Intraoperatively, the surgeon encountered three times vessel damage. In conclusion, a wide range of complications rates following TDR (1–90.0%) was found in all cohort studies. The majority of the studies reported complication rates ranging from 10 to 40%. Reoperation at index level was reported in 1.0–28.6%. The three randomized controlled trials published overall complication rates range from 7.2 to 28.6% in the TDR group and 6.7 to 50.2% in the fusion group. The overall reoperation rate at the index level ranged from 3.7 to 11.4% in the TDR group and 5.4–26.1% in the fusion group. However, much higher rates were reported in FDA reports on the Charité and ProDisc trials.
Discussion
In this article, we systematically reviewed the available literature on the clinical course, effectiveness, cost-effectiveness, and safety of TDR in patients with symptomatic DDD. Sixteen prospective cohort studies were identified that assessed the course of complaints and symptoms. These studies suggest pain relief, improvement in functional status and patient satisfaction after TDR. However, the quality of reporting on outcomes was often poor, hampering an adequate interpretation. In addition, a significant amount of complications was reported. These cohort studies lacked control group, which is necessary to evaluate effectiveness of TDR. Only three randomized controlled multicentre trials were identified that had assessed the effectiveness of TDR. The results show that there is low quality evidence (based on one study only with a low risk of bias) that there are no clinically relevant differences on the primary outcome measures between the Charité group and the BAK cage at 2 years follow up, and there is very low quality evidence (based on 1 study only with a high risk of bias) that there are no clinically relevant differences on the primary outcome measures at 5 years follow up. Furthermore, there is very low quality evidence (based on one study only with a high risk of bias) for contradictory results on the primary outcome measures for the ProDisc when compared with anterior lumbar circumferential fusion at the 2-year follow up. There is insufficient evidence on the Flexicore, because this trial had a high risk of bias, and should be considered as a preliminary report because it only reported on a small proportion of all included patients who participated in this multi centre trial.
For assessing the complication rate, all reported complications were extracted from the cohort studies and randomized controlled trials included in this review, as well as overview studies on complication rates. A wide range of complications rates following TDR (1–91.0%) was found in the cohort studies. The majority of the studies reported complication rates ranging from 10 to 40%. Reoperation at the index level was reported in 1.0–28.6%. In the three randomized controlled trials, overall complication rates ranged from 7.3 to 29.1% in the TDR group and from 6.3 to 50.2% in the fusion group. The overall reoperation rate at the index level ranged from 3.7 to 11.4% in the TDR group and from 5.4 to 26.1% in the fusion group. However, much higher rates were reported in FDA reports on the Charité and ProDisc trials. No full economic evaluation was identified, so there is no evidence regarding the costeffectiveness of TDR.
The course of DDD complaints and/or symptoms following total disc replacement surgery
We identified 16 prospective cohort studies to evaluate the course of DDD complaints and/or symptoms. The outcome results suggested a positive course after TDR with a high proportion of patients satisfied with the result. However, these studies were of poor methodological quality and detailed information on how outcomes were measured was often lacking. For example, it was often unclear which criteria were used for clinical success and how return to work was measured. Furthermore, another drawback is that a significant amount of complications was reported as well.
Moreover, these results have to be interpreted in light of controversy and limited literature regarding the causal relationship between DDD and chronic low back pain [4]. Boden et al. [66] reported on 67 asymptomatic individuals assessed for DDD with MRI. DDD was seen in 34% of the individuals between 20 and 39 years of age; 59% of individuals between 40 and 59 years of age, and in all but one (93%) between 60 and 80 years of age. Jensen et al. [67] reported on 98 asymptomatic people after MRI and concluded that 64% of these people had an intervertebral disc abnormality. This challenges the rationale of surgery for DDD in the absence of convincing pathological pathways of DDD.
The effectiveness of total disc replacement surgery compared to other treatments
The Flexicore trial should be interpreted with great caution because of the high risk of bias. Of the three randomized controlled trials, the 2-year follow up of the Charité trial was considered to have a low risk of bias. However, the fusion technique with BAK cages and the iliac crest bone autograft used in this trial are techniques that are no longer commonly used because of poor outcomes [68–70]. A better comparator would be the circumferential fusion technique which was used in the ProDisc and Flexicore trials. The use of autograft in all three studies may also be criticized as many surgeons now use both recombinant BMP-2 and/or percutaneous pedicle screw fixation when performing lumbar fusion [55]. Use of an inadequate control intervention brings into question the clinical relevance of the results of the three trials. An additional concern is the fact that the literature is still controversial about the superiority of fusion compared to conservative treatment [5, 15, 71, 72]. For this reason, it can be interesting to compare the effectiveness of TDR to conservative treatment. At present, no studies comparing total disc replacement surgery to other treatments have been published.
The three randomized controlled trials selected patients carefully, scrutinizing various contraindications for TDR. Because of this careful selection, the published trials do not provide evidence for the widespread use of TDR in all patients with DDD. The relevance of the clinical outcomes in the Charité and the ProDisc trials can also be challenged. First, modest success rates were observed in both the TDR and the fusion groups. In the Charité trial, only 57.1% of patients with TDR met all 4 criteria for success, when compared with 46.5% in the fusion group (P < 0.0001). In the ProDisc group, only 53.4% of patients with TDR met all 10 FDA criteria for success, when compared with 40.8% in the fusion group (P = 0.0438). Second, in the Prodisc trial, 69.1% of TDR subjects improved by more than 25% on the Oswestry, when compared with 54.9% in the fusion group. In the Charité trial, 63.9% of TDR subjects improved by more than 25% on the Oswestry, compared to 50.5% in the fusion group. The use of the 25% benchmark for improvement should be interpreted against a background of a recently published consensus statement that advocates a 30% improvement in Oswestry as a benchmark for clinically relevant improvement. This recommendation focussed primarily on conservative interventions in a primary care setting. It was suggested that it might be more appropriate to use larger change scores as benchmarks for expensive and risky procedures [73]. Third, one of the purposes of the device implementation is to reduce low back pain whereas the definition of success did not consider pain relief or opioid use. Finally, Oswestry and VAS cannot discriminate between pain that is residual from the iliac crest after fusion surgery versus the lumbar spine. Therefore, Oswestry and VAS may be artificially higher in the fusion group compared with TDR.
The ODI was used in all included RCT’s, but different versions of the ODI were used. Sasso used ODIv2.0 [53]. Blumenthal used the ODIv1.0 and Zigler used the ODI (chiropractic revised version [74]) [75]. Because different versions of the ODI are used, a direct comparison between studies is hampered. Zigler, however, holds the opinion that the differences between the various ODI versions are subtle and, they think, inconsequential [77].
Davidson [77] and Fairbanks [75] hold the opinion that the amendments of this ‘chiropractic revised version’ are major and therefore this version cannot be compared with the official versions of ODI.
The safety of the total disc replacement surgery
Complications have been poorly described in the prospective cohort studies and the randomized controlled trials. It is interesting that the complications rates and reoperation rates are lower in the published articles than in the FDA reports [56, 58]. This illustrates the complexity of reporting on adverse effects. Compared to the journals where the papers were published, apparently the FDA requires exhaustive and detailed reporting of “adverse events” most of which have no relationship to the success or failure of the prosthesis. Complications associated with lumbar fusion include incomplete relief of pain, loss of motion, loss of sagittal balance, pseudoarthrosis, adjacent segment degeneration, and bonegraft donor site complication. However, a separate set of concerns exist in TDR. Wear debris leading to osteolysis and systematic effects, vertebral body damage, posterior migration or extrusion may lead to device failure and serious vascular complications. Prosthesis that fail to adequately replicate the physiologic kinematics of the lumbar spine may predispose the patient to facet joint degeneration. Without true motion preservation, the devices will merely act as interbody spacers with no potential to prevent adjacent level degeneration [78]. Finally, reported complications for TDR show there can be severe and even life threatening, e.g. major vascular injury, major nerve root damage and device failure. However, these complication rates are low [9, 17, 79, 80].
Furthermore, in the two low risks of bias studies [10, 52], the re-operation rates in the TDR group are slightly higher than in the fusion group. However, this has to be balanced against the fact that re-operation procedures for TDR are more complex.
The use of intervertebral disc prostheses as an alternative to spinal fusion has been advocated to preserve segmental motion and to prevent adjacent degeneration. However, there is no consensus on this subject in literature. Some studies suggest adjacent level degeneration is prevented after TDR [6, 12]. However, other studies show adjacent disc degeneration after TDR [61, 81]. This could be the result of the DDD itself, spreading to multiple levels of the spine, and/or be a consequence of stresses on adjacent levels, generated from unphysiological motion and functioning of the disc prosthesis [61]. Moreover, there is little knowledge regarding complications on the long term. Putzier et al. [81] published a retrospective study with 17 years follow-up and reoperation was necessary in 11% of patients. It is important to know more about long-term complications because most operated patients are of relatively young age, between 30 and 50 years. A disc prosthesis used for TDR should survive for at least 40 years. It is very questionable if the lifetime of the designs now available will be that long as little is known about long-term behaviour of biomaterials in the spine. We do know that revision surgery can be dangerous because of adherence to great vessels and the nerve plexus. Studies that review long-term complications and longevity of the prostheses are highly recommended.
Conclusion
There is low quality evidence that there are no clinically relevant differences on the primary outcome measures between the Charité group and the BAK cage at 2 years follow up, and there is very low quality evidence that there are no clinical relevant differences on the primary outcome measures at 5 years follow up. For the ProDisc device, there is very low quality evidence for contradictory results on the primary outcome measures when compared with anterior lumbar circumferential fusion. Furthermore, reported complication rates varied from 1.0 to 91.0% in cohort studies and 7.3 to 29.1% in randomized controlled trials. Still lacking are high quality prospective, controlled, long-term follow-up studies, including a full economic evaluation taking into account all relevant cost when compared with the clinical benefit, and with relevant control groups to establish the efficiency and the longevity of the devices. The existing evidence, specifically regarding long-term effectiveness and/or safety is considered insufficient to justify the widespread use of TDR over fusion for single level degenerative disc. It is recommended that disc replacement surgery at this time only is performed within prospective scientific studies until further documentation of its efficiency is provided.
References
Van Tulder MW, Koes BW, Bouter LM (1995) A cost-of-illness study of back pain in The Netherlands. Pain 1995:233–240
Pengel LHM, Herbert RD, Maher CG et al (2003) Acute low back pain: systematic review of its prognosis. BMJ 327:323
Errico TJ (2005) Lumbar disc arthroplasty. Clin Orthop Relat Res 435:106–117
Frelinghuysen P, Huang RC, Girardi FP et al (2005) Lumbar total disc replacement part I: rationale, biomechanics, and implant types. Orthop Clin North Am 36:293–299
Fritzell P, Hagg O, Wessberg P et al (2001) Lumbar fusion versus nonsurgical treatment for chronic low back pain: a multicenter randomized controlled trial from the Swedish Lumbar Spine Study Group. Spine 26:2521–2532
Hochschuler SH, Ohnmeiss DD, Guyer RD et al (2002) Artificial disc: preliminary results of a prospective study in the United States. Eur Spine J 2:S106–S110
Errico TJ (2004) Why a mechanical disc? Spine J 4:151S–157S
Freeman BJC, Davenport J (2006) Total disc replacement in the lumbar spine: a systematic review of the literature. Eur Spine J 15(Suppl 3):S439–S447
de Kleuver M, Oner FC, Jacobs WC (2003) Total disc replacement for chronic low back pain: background and a systematic review of the literature. Eur Spine J 12:S108–S116
Blumenthal S, McAfee PC, Guyer RD et al (2005) A prospective, randomized, multicenter Food and Drug Administration investigational device exemptions study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: part I: evaluation of clinical outcomes. Spine 30:1565–1575
Cunningham BW, Dmitriev AE, Hu N et al (2003) General principles of total disc replacement arthroplasty: seventeen cases in a nonhuman primate model. Spine 28:S118–S124
Link HD (2002) History, design and biomechanics of the LINK SB Charite artificial disc. Eur Spine J 11:S98–S105
McAfee PC, Cunningham B, Holsapple G et al (2005) A prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: part II: evaluation of radiographic outcomes and correlation of surgical technique accuracy with clinical outcomes. Spine 30:1576–1583
Szpalski M, Gunzburg R, Mayer M (2002) Spine arthroplasty: a historical review. Eur Spine J 11(Suppl 2):S65–S84
Fairbank J, Frost H, Wilson-MacDonald J et al (2005) Randomised controlled trial to compare surgical stabilisation of the lumbar spine with an intensive rehabilitation programme for patients with chronic low back pain: the MRC spine stabilisation trial. BMJ 330:1233
Fernstrom U (1966) Arthroplasty with intercorporal endoprothesis in herniated disc and in painful disc. Acta Chir Scand Suppl 357:154–159
Randolph GB, Scioscia TN, Wang JC (2006) Lumbar total disc arthroplasty: state of the data. Semin Spine Surg 18:61–71
van Tulder M, Furlan A, Bombardier C et al (2003) Updated method guidelines for systematic reviews in the Cochrane collaboration back review group. Spine 28:1290–1299
Brozek J, Akl E, Alonso-Coello P, on behalf of the GRADE Working Group et al (2009) Grading quality of evidence and strength of recommendations in clinical practice guidelines. Allergy 64:669–677
Lemaire JP, Skalli W, Lavaste F et al (1997) Intervertebral disc prosthesis. Results and prospects for the year 2000. Clin Orthop Relat Res 337:64–76
Zeegers WS, Bohnen LM, Laaper M et al (1999) Artificial disc replacement with the modular type SB Charite III: 2-year results in 50 prospectively studied patients. Eur Spine J 8:210–217
Regan JJ (2005) Clinical results of charite lumbar total disc replacement. Orthop Clin North Am 36:323–340
Lemaire JP, Carrier H, Eh SariAli et al (2005) Clinical and radiological outcomes with the Charite artificial disc: a 10-year minimum follow-up. J Spinal Disord Tech 18:353–359
Ross R, Mirza AH, Norris HE et al (2007) Survival and clinical outcome of SB Charite III disc replacement for back pain. J Bone Joint Surg Br 89:785–789
Gioia G, Mandelli D, Randelli F (2007) The Charite III artificial disc lumbar disc prosthesis: assessment of medium-term results. J Orthop Traumatol 8:134–139
Warachit P (2008) Results of Charite artificial lumbar disc replacement: experience in 43 Thais. J Med Assoc Thai 91:1212–1217
Bertagnoli R, Kumar S (2002) Indications for full prosthetic disc arthroplasty: a correlation of clinical outcome against a variety of indications. Eur Spine J 11(Suppl 2):S131–S136
Tropiano P, Huang RC, Girardi FP et al (2003) Lumbar disc replacement: preliminary results with ProDisc II after a minimum follow-up period of 1 year. J Spinal Disord Tech 16:362–368
Tropiano P, Huang RC, Girardi FP et al (2005) Lumbar total disc replacement. Seven to eleven-year follow-up. J Bone Joint Surg Am 87:490–496
Bertagnoli R, Yue JJ, Shah RV et al (2005) The treatment of disabling single-level lumbar discogenic low back pain with total disc arthroplasty utilizing the Prodisc prosthesis: a prospective study with 2-year minimum follow-up. Spine 30:2230–2236
Bertagnoli R, Yue JJ, Shah RV et al (2005) The treatment of disabling multilevel lumbar discogenic low back pain with total disc arthroplasty utilizing the ProDisc prosthesis: a prospective study with 2-year minimum follow-up. Spine 30:2192–2199
Chung SS, Lee CS, Kang CS (2006) Lumbar total disc replacement using ProDisc II: a prospective study with a 2-year minimum follow-up. J Spinal Disord Tech 19:411–415
Siepe CJ, Mayer HM, Wiechert K et al (2006) Clinical results of total lumbar disc replacement with ProDisc II: three-year results for different indications. Spine 31:1923–1932
Siepe CJ, Mayer HM, Heinz-Leisenheimer M et al (2007) Total lumbar disc replacement: different results for different levels. Spine 32:782–790
Ogon M, Howanietz N, Tuschel A et al (2007) Implantation of the Prodisc® intervertebral disk prosthesis for the lumbar spine. Oper Orthop Traumatol 19:209–230
Mayer HM, Wiechert K, Korge A et al (2002) Minimally invasive total disc replacement: surgical technique and preliminary clinical results. Eur Spine J 11(Suppl 2):S124–S130
Le Huec JC, Mathews H, Basso Y et al (2005) Clinical results of Maverick lumbar total disc replacement: two-year prospective follow-up. Orthop Clin North Am 36:315–322
Le Huec JC, Basso Y, Aunoble S et al (2005) Influence of facet and posterior muscle degeneration on clinical results of lumbar total disc replacement: two-year follow-up. J Spinal Disord Tech 18:219–223
Le Huec J, Basso Y, Mathews H et al (2005) The effect of single-level, total disc arthroplasty on sagittal balance parameters: a prospective study. Eur Spine J 14:480–486
Fraser RD, Ross ER, Lowery GL et al (2004) AcroFlex design and results. Spine J 4:245S–251S
McAfee PC, Fedder IL, Saiedy S et al (2003) Experimental design of total disk replacementexperience with a prospective randomized study of the SB Charite. Spine 28:S153–S162
McAfee PC, Fedder IL, Saiedy S et al (2003) SB Charite disc replacement: report of 60 prospective randomized cases in a US center. J Spinal Disord Tech 16:424–433
Guyer RD, McAfee PC, Hochschuler SH et al (2004) Prospective randomized study of the Charite artificial disc: data from two investigational centers. Spine J 4:252S–259S
Blumenthal SL, Ohnmeiss DD, Guyer RD et al (2003) Prospective study evaluating total disc replacement: preliminary results. J Spinal Disord Tech 16:450–454
Zigler JE (2004) Lumbar spine arthroplasty using the ProDisc II. Spine J 4:260S–267S
Delamarter RB, Fribourg DM, Kanim LEA et al (2003) ProDisc artificial total lumbar disc replacement: introduction and early results from the United States clinical trial. Spine 28:S167–S175
Zigler JE (2003) Clinical results with ProDisc: European experience and U.S. investigation device exemption study. Spine 28:S163–S166
Zigler JE, Burd TA, Vialle EN et al (2003) Lumbar spine arthroplasty: early results using the ProDisc II: a prospective randomized trial of arthroplasty versus fusion. J Spinal Disord Tech 16:352–361
Delamarter RB, Bae HW, Pradhan BB (2005) Clinical results of ProDisc-II lumbar total disc replacement: report from the United States clinical trial. Orthop Clin North Am 36:301–313
Auerbach JD, Wills BPD, McIntosh TC et al (2005) Lumbar disc arthroplasty versus fusion for single-level degenerative disc disease: two-year results from a randomized prospective study. Semin Spine Surg 17:310–318
Guyer RD, McAfee PC, Banco RJ et al (2009) Prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: five-year followup. Spine J 9:374–386
Zigler J, Delamarter R, Spivak JM et al (2007) Results of the prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of the ProDisc-L total disc replacement versus circumferential fusion for the treatment of 1-level degenerative disc disease. Spine 32:1155–1162
Sasso RC, Foulk DM, Hahn M (2008) Prospective, randomized trial of metal-on-metal artificial lumbar disc replacement: initial results for treatment of discogenic pain. Spine 33:123–131
Mayer HM, Siepe C (2007) Total lumbar disc arthroplasty. Curr Orthop 21:17–24
German JW, Foley KT (2005) Disc arthroplasty in the management of the painful lumbar motion segment. Spine 30:S60–S67
FDA approval Charité: http://www.fda.gov/cdrh/pdf4/P040006a.pdf
McAfee PC, Geisler FH, Saiedy SS et al (2006) Revisability of the CHARITE artificial disc replacement: analysis of 688 patients enrolled in the U.S. IDE study of the CHARITE Artificial Disc. Spine 31:1217–1226
FDA approval ProDisc: http://www.fda.gov/cdrh/pdf5/p050010a.pdf
Geisler FH, Blumenthal SL, Guyer RD et al (2004) Neurological complications of lumbar artificial disc replacement and comparison of clinical results with those related to lumbar arthrodesis in the literature: results of a multicenter, prospective, randomized investigational device exemption study of Charite intervertebral disc. J Neurosurg Spine 1:143–154
Leary SP, Regan JJ, Lanman TH et al (2007) Revision and explantation strategies involving the CHARITE lumbar artificial disc replacement. Spine 32:1001–1011
Punt IM, Visser VM, van Rhijn LW et al (2008) Complications and reoperations of the SB Charite lumbar disc prosthesis: experience in 75 patients. Eur Spine J 17:36–43
van Ooij A, Oner FC, Verbout AJ (2003) Complications of artificial disc replacement: a report of 27 patients with the SB Charite disc. J Spinal Disord Tech 16:369–383
van Ooji A, Schurink GW, Oner FC et al (2007) Findings in 67 patients with recurrent or persistent symptoms after implantation of a disc prosthesis for low back pain. Ned Tijdschr Geneeskd 151:1577–1584
Kurtz SM, van Ooij A, Ross R et al (2007) Polyethylene wear and rim fracture in total disc arthroplasty. Spine J 7:12–21
van Ooij A, Kurtz SM, Stessels F et al (2007) Polyethylene wear debris and long-term clinical failure of the Charite disc prosthesis: a study of 4 patients. Spine 32:223–229
Boden SD, Davis DO, Dina TS et al (1990) Abnormal magnetic-resonance scans of the lumbar spine in asymptomatic subjects. A prospective investigation. J Bone Joint Surg Am 72:403–408
Jensen MC, Brant-Zawadzki MN, Obuchowski N et al (1994) Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med 331:69–73
Button G, Gupta M, Barrett C et al (2005) Three- to six-year follow-up of stand-alone BAK cages implanted by a single surgeon. Spine J 5:155–160
Lorenz M, Zindrick M, Schwaegler P et al (1991) A comparison of single-level fusions with and without hardware. Spine 16:S455–S458
Pellise F, Puig O, Rivas A et al (2002) Low fusion rate after L5–S1 laparoscopic anterior lumbar interbody fusion using twin stand-alone carbon fiber cages. Spine 27:1665–1669
Brox JI, Reikeras O, Nygaard O et al (2006) Lumbar instrumented fusion compared with cognitive intervention and exercises in patients with chronic back pain after previous surgery for disc herniation: a prospective randomized controlled study. Pain 122:145–155
Van Tulder MW, Koes B, Seitsalo S et al (2006) Outcome of invasive treatment modalities on back pain and sciatica: an evidence-based review. Eur Spine J 15:S82–S92
Ostelo RWJG, Deyo RA, Stratford P et al (2008) Interpreting change scores for pain and functional status in low back pain: towards international consensus regarding minimal important change. Spine 33:90–94
Hudson-Cook N, Tomes-Nicholson K, Breen A (1989) A revised Oswestry disability questionnaire. In: Roland M, Jenner JR (eds) Back pain: new approaches to rehabilitation and education. Manchester University Press, Manchester, pp 187–204
Fairbank J (2007) Letter to the Editor. Spine 32(25):2929–2930
Stieber JR, Donald GD (2006) Early failure of lumbar disc replacement: case report and review of the literature. J Spinal Disord Tech 19:55–60
Zigler JE (2007) Letter to the editor. Spine 32(25):2930–2931
Brau SA (2002) Mini-open approach to the spine for anterior lumbar interbody fusion: description of the procedure, results and complications. Spine J 2:216–223
Bertagnoli R, Zigler J, Karg A et al (2005) Complications and strategies for revision surgery in total disc replacement. Orthop Clin North Am 36:389–395
Harrop JS, Youssef JA, Maltenfort M et al (2008) Lumbar adjacent segment degeneration and disease after arthrodesis and total disc arthroplasty. Spine 33:1701–1707
Putzier M, Funk JF, Schneider SV et al (2006) Charite total disc replacement—clinical and radiographical results after an average follow-up of 17 years. Eur Spine J 15:183–195
Acknowledgments
For this review, the authors received a grant from The Health Care Insurance Board (CVZ), Diemen, The Netherlands.
Conflict of interest statement
For this review, the authors received a Grant from The Health Care Insurance Board (CVZ), Dieman, The Netherlands.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van den Eerenbeemt, K.D., Ostelo, R.W., van Royen, B.J. et al. Total disc replacement surgery for symptomatic degenerative lumbar disc disease: a systematic review of the literature. Eur Spine J 19, 1262–1280 (2010). https://doi.org/10.1007/s00586-010-1445-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00586-010-1445-3