
Abstract
CHARGE syndrome (OMIM 214800) is a rare autosomal-dominant congenital malformation syndrome that results from haploinsufficiency of the chromodomain helicase DNA-binding protein 7 (CHD7). We performed a phenotypic characterization and genetic analysis of CHD7 in 18 Korean patients with CHARGE syndrome. Eighteen unrelated Korean patients (10 females and 8 males; age range 0.0–19.6 years) with CHARGE syndrome were enrolled. Clinical data were collected by retrospective review of medical records. A serial analysis via sequencing and multiple ligation-dependent probe amplification of CHD7 was performed to determine the molecular genetic spectrum of the patients. The prevalence of cardinal symptoms was as follows: coloboma (13/18, 72.2%), heart defects (13/18, 72.2%), choanal atresia/stenosis (4/18, 22.2%), retarded growth (10/18, 55.6%), genital anomalies (15/18, 83.3%) and ear abnormalities (18/18, 100%). Five patients had cerebellar vermis hypoplasia (5/17, 29.4%) with no clinical symptoms or signs of cerebellar dysfunction. Furthermore, we identified genetic alterations in all 18 patients, including 10 novel mutations. Considering its frequency among patients with CHD7 mutations, cerebellar vermis hypoplasia may be a clinical diagnostic clue of CHARGE syndrome, although it is not included in the diagnostic critieria. And, the identification of CHD7 mutations may help the confirmative diagnosis.
Similar content being viewed by others

Introduction
CHARGE syndrome (OMIM 214800) is a rare autosomal-dominant congenital malformation that is estimated to occur in 1:10 000 births worldwide.1 Although Pagon et al.2 originally summarized six cardinal clinical symptoms of CHARGE syndrome, including coloboma of the eye, heart defects, atresia of choanae, retardation of growth and development, genital hypoplasia and ear anomalies, the phenotype of this syndrome is highly heterogeneous and shows variable combinations of these symptoms. Moreover, to date, the clinical diagnostic criteria have been revised several times.2, 3, 4 Cerebellar vermis hypoplasia is not traditionally included in the diagnostic criteria of CHARGE syndrome, even though a higher mortality rate was reported in patients with central nervous system malformation, including ventriculomegaly and brain stem/cerebellar anomalies.5
In 2004, it was demonstrated that CHARGE syndrome results from haploinsufficiency of the chromodomain helicase DNA-binding protein 7 (CHD7).6 CHD7 has a critical role in the formation, acquisition of differentiation and migration throughout the body of the migratory neural crest, and which gives rise to craniofacial bone cartilages, the peripheral nervous system and cardiac structures.7, 8, 9, 10 Heterozygous mutation or deletion of CHD7 results in alteration of gene expression, DNA repair and replication and recombination, and has important roles in embryonic development.6 Heterozygous mutation of CHD7 is responsible for ~90% of typical patients fulfilling clinical diagnostic criteria of CHARGE syndrome.7, 11, 12, 13, 14
In Korea, several case reports and small case series of genetically confirmed CHARGE syndrome have been reported.15, 16, 17, 18, 19 In this study, we performed a phenotypic characterization and genetic analysis of the CHD7 gene in 18 unrelated Korean patients with CHARGE syndrome.
Materials and Methods
Patients and clinical data analysis
Eighteen unrelated Korean patients (10 females and 8 males; age range 0.0–19.6 years) with CHARGE syndrome, from two different clinical genetics centers in Korea, were enrolled in the study from May 2010 to May 2015. All patients met at least two of the three major signs of CHARGE syndrome, as defined by Verloes.4 At the time of enrollment, complete physical and neurologic examination was performed by the clinical geneticists. Medical history of the patients was collected by retrospective review of medical records. Physical growth was evaluated based on Korean reference data20 and expressed as a standard deviation score (SDS). Developmental delay was assessed using the Bayley Scales of Infant Development II or Korean-Wechsler Preschool and Primary Scales of Intelligence (K-WPPSI) or Korean-Wechsler Intelligence Scales for Children (K-WISC) or Korean-Wechsler Adult Intelligence Scale (K-WAIS) according to the age of the patients. Presence of hypogonadotrophic hypogonadism was assessed by gonadotrophin-releasing hormone stimulation test. Anosmia was assessed by Korean Version of Sniffin’ Sticks (KVSS) test, who can perform and agree with the test. For the others, it was assessed by the clinical inquiry. Written consent was obtained from all participants who provided identifiable samples. This study was approved by the Institutional Review Board of the Seoul National University Hospital and Ajou University Hospital.
Genetic analysis
A sequencing analysis of CHD7 was performed first. In cases with no sequence variation, multiple ligation-dependent probe amplification (MLPA) was performed to identify the deletion/duplication or whole-gene deletion of CHD7.
Sequencing analysis of CHD7
Blood specimens were collected from patients and their parents. Genomic DNA was isolated from peripheral blood leukocytes using a Wizard Genomic DNA Purification Kit, according to the manufacturer’s instructions (Promega, Madison, WI, USA). All exons (1–38) and exon–intron boundaries of CHD7 (reference sequence: NG_007009.1 and NM_017780.3) were amplified by polymerase chain reaction using appropriate primers that were designed by the authors (available on request) and a thermal cycler (Applied Biosystems, Foster City, CA, USA). Direct sequencing was performed using a BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems), and the analysis was conducted using an ABI3130xl Genetic Analyzer (Applied Biosystems).
MLPA
MLPA analyses were performed using the SALSA P201 Kit for CHARGE syndrome (MRC Holland, Amsterdam, The Netherlands), according to the manufacturer’s instructions. Amplified products were separated on an ABI3130xl Genetic Analyzer (Applied Biosystems) and analyzed using the Gene Mapper Software (Applied Biosystems).
Results
Clinical spectrum
Eighteen unrelated Korean patients with clinically and genetically confirmed CHARGE syndrome were analyzed in this study. The clinical features of these patients are shown in Table 1. The prevalence of the observed clinical features is shown in Figure 1.

The prevalence of clinical features of CHARGE syndrome found in this study comparing with the previous report. The overall prevalence of each clinical feature observed in this study (black bar) is in accord with those of previous report (white bar).14
All 18 patients had external ear anomalies and hearing loss. The second most frequent symptom was developmental delay (17/18), followed by genital anomalies (15/18) and facial nerve palsy (14/18). IX or X cranial nerve palsy (13/18), coloboma (13/18) and heart defects (13/18) were other frequently observed clinical features. Growth retardation was present in 10 patients.
Coloboma and microphthalmia
Thirteen (72.2%) of the 18 patients had coloboma of the eye. Among these, 3 patients had unilateral coloboma and 10 patients had bilateral coloboma. Five patients had coloboma in multiple locations. Regarding the location of ocular coloboma, all 13 patients had chorioretinal coloboma and no iris coloboma was found. Among the chorioretinal coloboma, choroid coloboma was observed most frequently (8/13 patients), followed by optic disc coloboma (5/13) and retinal coloboma (3/13). Seven patients (38.9%) had microphthalmia, including one case of bilateral and six cases of unilateral involvement.
Heart defects
Heart defects were presented in 13 patients (72.2%). Among them, 11 patients underwent surgical or interventional correction. The patients had variable combinations of heart anomalies. The types of heart defects were: 11 cases (patient 2, 3, 6, 7, 9, 11, 12, 13, 14, 16 and 17) of patent ductus arteriosus (PDA), seven cases (patient 1, 2, 10, 11, 13, 16 and 17) of atrial septal defect, one case (patient 1) of ventricular septal defect and one case (patient 9) of aortic stenosis (AS). There was no patient with mid-line heart defect including tetraology of Fallot.
Choanal atresia/stenosis
Choanal atresia was observed in three patients. One patient had unilateral choanal atresia and the remaining two patients had bilateral involvement. All three patients with choanal atresia had undergone surgical correction. Choanal stenosis was found in one patient.
Retarded growth and developmental delay
Two patients were born as preterm neonates, at 35 weeks of gestation. Among the remaining 16 patients, 3 had intrauterine growth retardation and were born as SGA. Postnatal growth retardation (height SDS ⩽–2.5 SDS or weight ⩽–2.5 SDS) was observed in 10 patients. The median weight z-score was –2.1 (range, −4.9 to −0.9) and the median height z-score was –2.6 (range –6.7 to –0.2). Developmental delay was observed in all but one of the patients.
Genital anomalies
Fifteen out of the 18 patients had genital anomalies. Among the 8 male patients, 5 had cryptorchidism and 2 had micropenis, whereas 9 out of the 10 female patients had hypoplastic labia.
All three patients of pubertal or postpubertal age (patients 4, 5 and 18) showed delayed puberty that resulted from hypogonadotrophic hypogonadism, which was assessed by gonadotrophin-releasing hormone stimulation test.
Ear anomalies
All 18 patients enrolled in this study had external ear anomalies. The anomalies reported included low-set, cup-formed, and ‘snipped-off’’ ears. Abnormal helix and microtia were also present.
Temporal bone CT or MRI was available for 13 patients, who all exhibited an absent or hypoplastic semicircular canal. Middle ear ossicular malformations were observed in 7 patients, and an inner ear Mondini defect of the cochlea was found in 8 patients.
All 18 patients had hearing loss; 15 out of the 18 patients had both sensorineural and conductive hearing loss. Among the remaining 3 patients, 2 patients had sensorineural-type hearing loss and 1 patient had conductive hearing loss. Nine patients underwent cochlear implantation surgery, and 5 patients had a hearing aid.
Central nervous system anomalies and cranial nerve dysfunction
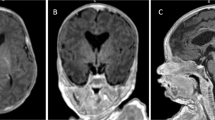
Cerebellar vermis hypoplasia and enlarged cisterna magna were found in 5 patients among the 17 patients (29.4%) for whom an MRI scan was available. Figure 2 shows a representative brain MR image. As described above, sensorineural hearing loss was found in 17 out of the 18 patients. Fourteen out of the 18 patients had VII (facial nerve) palsy, and 13 out of the 18 patients had IX/X nerve dysfunction, accompanied by swallowing problems or dysarthria. Anosmia was noted in 2 of 4 patients performed Korean version of Sniffin’ Sticks test, and both of these patients were also associated with Kallmann syndrome.

The brain MR images of patients with cerebellar vermis hypoplasia. The brain MR images show cerebellar vermis hypoplasia in patient 2 (a), patient 4 (b), patient 8 (c) and patient 16 (d), respectively.
Other manifestations
Five patients had tracheostomy because of extubation failure resulting from laryngomalacia (4/5 patients) and glottis stenosis (1/5 patients). Urogenital anomalies were noted in 2 patients, both of whom had vesicoureteral reflux. Five patients had skeletal anomalies, including scoliosis (3 patients), clinodactyly (1 patient) and thumb dysplasia (1 patient). Two patients had a hiatal hernia and underwent surgical correction. None of our patients had a tracheoesophageal fistula.
Genetic spectrum of CHD7
We found a genetic alteration of the CHD7 gene in all 18 patients, all of which were de novo mutations. Sixteen sequence variants were found by sequencing analysis, and two deletions were detected by MLPA analysis. The results pertaining to the genetic alterations detected in the patients are described in Table 2. All identified mutations were unique and scattered throughout the whole CHD7 gene. Among these mutations, 10 (55.6%) were novel, whereas the remaining eight mutations had been reported previously (Table 2). In this study, nonsense mutations were by far the most frequently detected alterations (8/18, 44.4%), followed by splice site mutations (4/18, 22.2%), and frameshift mutations (3/18, 16.7%). Large exonic deletions were found in 2 patients (11.1%). One patient (patient 2) had a missense mutation (p.Gln1214Arg).
Discussion
In this study, we described the detailed clinical features of 18 Korean patients with CHARGE syndrome and identified the molecular genetic spectrum of the CHD7 gene, which included 10 novel mutations in these 18 unrelated Korean patients with CHARGE syndrome.
Four features are almost always present in patients with CHD7 mutation: external ear anomalies, cranial nerve dysfunction, semicircular canal hypoplasia, and delayed motor milestones.14 These findings were consistently present in our cohort (Figure 1). All 18 patients had external ear anomalies and hearing loss, which is consistent with previous reports.11 In this study, the second most frequent symptom was developmental delay (17/18), followed by genital anomalies (15/18) and facial nerve palsy (14/18). Therefore, a full systemic examination of patients with external ear anomalies and hearing loss may be helpful for identifying patients with CHARGE syndrome.
Although many clinical features are shared by other multiple anomaly syndromes, including 22q11.2 deletion, Kallmann syndrome and Treacher Collins syndrome, CHARGE syndrome is considered unique in its combination of distinctive inner and outer ear defects and optic colobomas.11, 21 Despite the fact that choanal atresia or stenosis is one of the characteristic clinical features of CHARGE syndrome, these findings were found in only four patients (22.2%) in our cohort, while the prevalence was reported 55% in the previous report14 (Figure 1). Therefore, the diagnosis of CHARGE syndrome should be considered even if patients do not have choanal atresia/stenosis.
In our cohort, among the 17 patients for whom brain MRI was available, cerebellar vermis hypoplasia was noted in 5 patients (29.4%), despite the fact that it is not included in the clinical diagnostic criteria of CHARGE syndrome. CHD7 controls the expression of other genes during embryonic development. Mutation of CHD7 causes various developmental anomalies, including neural defects. Yu et al.22 recently reported a variable degree of cerebellar vermis hypoplasia and consequential enlarged fluid-filled spaces surrounding the cerebellum in 35% (7/20) of patients with CHARGE syndrome. Moreover, those authors also demonstrated that reduced FGF8 expression, which is a critical signal for early cerebellar development, results from CHD7 haploinsufficiency and is responsible for cerebellar vermis hypoplasia.22 Although our patients with cerebellar vermis hypoplasia did not show definite cerebellar dysfunction, such as gait disturbance or ataxia, neurological investigations including thorough neurologic exam and brain MRI is warranted to identify cerebellar vermis hypoplasia. However, our cohort is too small to draw a definite conclusion for the high frequency of cerebellar vermis hypoplasia in Korean patients with CHARGE syndrome. Further evaluation in large cohort is warranted for the certain conclusion. Furthermore, as this study is cross-sectional, the longitudinal follow-up study is also warranted because our cohort includes very young children <1 year.
As exonic copy-number alterations of the CHD7 gene are not a major cause of CHARGE syndrome,23 for molecular testing, sequencing analysis of CHD7 confirmed the diagnosis in the majority of cases in this study. If no mutation was identified by sequencing analysis, deletion/duplication analysis by MLPA was performed to identify large exonic deletions or whole-gene deletions. Using this sequential genetic analysis strategy, we were able to identify CHD7 mutations in all 18 patients. It is well known that truncating mutations are found in most patients, and that missense mutations are found in a minority of patients,11, 14 which was confirmed in this study, as only one patient (patient 2) in our cohort had a missense mutation.
To date, about 700 different pathogenic mutations have been reported (http://www.hgmd.cf.ac.uk). Most mutations are unique, and recurrent mutations are rare.11 As the clinical manifestations of patients with CHARGE syndrome are highly variable, even patients with the same mutation can exhibit different clinical phenotypes.12 Despite the fact that patients with missense mutations show a relatively milder clinical manifestation than do patients with truncating mutations,11 the only patient in this study who carried a missense mutation (patient 2) had a prenatal history of polyhydramnios and cisterna magna, as observed by fetal ultrasonography, and showed severe clinical manifestations, including laryngomalacia that required tracheostomy, hydronephrosis with bilateral high-grade vesicoureteral reflux, severe feeding intolerance that required percutaneous gastrostomy, bilateral facial nerve palsies, bilateral cryptorchidism, sensorineural hearing loss that required hearing aids, cleft palate and severely delayed cognitive and motor development. Therefore, it was difficult to identify clear genotype–phenotype correlations. In conclusion, we describe the clinical and genetic spectrum of 18 Korean patients with CHARGE syndrome, and identified 10 novel mutations of the CHD7 gene. Cerebellar vermis hypoplasia, which has not been a noticeable sign of CHARGE syndrome,5 is present in ~30% of patients in the cohort. Considering the frequency, cerebellar vermis hypoplasia may be a clinical diagnostic clue of CHARGE syndrome. Moreover, we suggest that physicians pay attention to the presence of cerebellar dysfunction among these patients during patient follow-up. And, the identification of CHD7 mutations may help the confirmative diagnosis.
References
Blake, K. D. & Prasad, C. CHARGE syndrome. Orphanet J. Rare Dis. 1, 34 (2006).
Pagon, R. A., Graham, J. M. Jr ., Zonana, J. & Yong, S. L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 99, 223–227 (1981).
Blake, K. D., Davenport, S. L., Hall, B. D., Hefner, M. A., Pagon, R. A., Williams, M. S. et al. CHARGE association: an update and review for the primary pediatrician. Clin. Pediatr. 37, 159–173 (1998).
Verloes, A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am. J. Med. Genet. Part A 133A, 306–308 (2005).
Issekutz, K. A., Graham, J. M. Jr ., Prasad, C., Smith, I. M. & Blake, K. D. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am. J. Med. Genet. Part A 133A, 309–317 (2005).
Vissers, L. E., van Ravenswaaij, C. M., Admiraal, R., Hurst, J. A., de Vries, B. B., Janssen, I. M. et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957 (2004).
Husu, E., Hove, H. D., Farholt, S., Bille, M., Tranebjaerg, L., Vogel, I. et al. Phenotype in 18 Danish subjects with genetically verified CHARGE syndrome. Clin. Genet. 83, 125–134 (2013).
Bajpai, R., Chen, D. A., Rada-Iglesias, A., Zhang, J., Xiong, Y., Helms, J. et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962 (2010).
Layman, W. S., Hurd, E. A. & Martin, D. M. Chromodomain proteins in development: lessons from CHARGE syndrome. Clin. Genet. 78, 11–20 (2010).
Schnetz, M. P., Handoko, L., Akhtar-Zaidi, B., Bartels, C. F., Pereira, C. F., Fisher, A. G. et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 6, e1001023 (2010).
Zentner, G. E., Layman, W. S., Martin, D. M. & Scacheri, P. C. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. Part A 152A, 674–686 (2010).
Jongmans, M. C., Admiraal, R. J., van der Donk, K. P., Vissers, L. E., Baas, A. F., Kapusta, L. et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 43, 306–314 (2006).
Lalani, S. R., Safiullah, A. M., Fernbach, S. D., Harutyunyan, K. G., Thaller, C., Peterson, L. E. et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 78, 303–314 (2006).
Bergman, J. E., Janssen, N., Hoefsloot, L. H., Jongmans, M. C., Hofstra, R. M. & van Ravenswaaij-Arts, C. M. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J. Med. Genet. 48, 334–342 (2011).
Cho, H. J., Song, M. H., Choi, S. Y., Kim, J., Lee, J., Kim, U. K. et al. Genetic analysis of the CHD7 gene in Korean patients with CHARGE syndrome. Gene 517, 164–168 (2013).
Lee, Y. W., Kim, S. C., Shin, Y. L., Kim, J. W., Hong, H. S., Lee, Y. K. et al. Clinical and genetic analysis of the CHD7 gene in Korean patients with CHARGE syndrome. Clin. Genet. 75, 290–293 (2009).
Song, M. H., Cho, H. J., Lee, H. K., Kwon, T. J., Lee, W. S., Oh, S. et al. CHD7 mutational analysis and clinical considerations for auditory rehabilitation in deaf patients with CHARGE syndrome. PLoS ONE 6, e24511 (2011).
Kim, Y., Lee, H. S., Yu, J. S., Ahn, K., Ki, C. S. & Kim, J. Identification of a novel mutation in the CHD7 gene in a patient with CHARGE syndrome. Korean J. Pediatr. 57, 46–49 (2014).
Lee, S. J., Chae, J. H., Lee, J. A., Cho, S. I., Seo, S. H., Park, H. et al. Non-homologous end joining repair mechanism-mediated deletion of CHD7 gene in a patient with typical CHARGE syndrome. Ann. Lab. Med. 35, 141–145 (2015).
Moon, J. S., Lee, S. Y., Nam, C. M., Choi, J. M., Choe, B. K., Seo, J. W. et al. 2007 Korean National Growth Charts: review of developmental process and an outlook. Korean. J. Pediatr. (Korean) 51, 1–25 (2008).
McMain, K., Blake, K., Smith, I., Johnson, J., Wood, E., Tremblay, F. et al. Ocular features of CHARGE syndrome. J. AAPOS 12, 460–465 (2008).
Yu, T., Meiners, L. C., Danielsen, K., Wong, M. T., Bowler, T., Reinberg, D. et al. Deregulated FGF and homeotic gene expression underlies cerebellar vermis hypoplasia in CHARGE syndrome. eLife 2, e01305 (2013).
Bergman, J. E., de Wijs, I., Jongmans, M. C., Admiraal, R. J., Hoefsloot, L. H. & van Ravenswaaij-Arts, C. M. Exon copy number alterations of the CHD7 gene are not a major cause of CHARGE and CHARGE-like syndrome. Eur. J. Med. Genet. 51, 417–425 (2008).
Acknowledgements
We thank the patients and their families for participating in this study. This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120030).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Sohn, Y., Ko, J., Shin, C. et al. Cerebellar vermis hypoplasia in CHARGE syndrome: clinical and molecular characterization of 18 unrelated Korean patients. J Hum Genet 61, 235–239 (2016). https://doi.org/10.1038/jhg.2015.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.135
This article is cited by
-
Phenotypic characteristics and variability in CHARGE syndrome: a PRISMA compliant systematic review and meta-analysis
Journal of Neurodevelopmental Disorders (2022)
-
ALS and CHARGE syndrome: a clinical and genetic study
Acta Neurologica Belgica (2018)
