




Abstract
We have recently reported brain atrophy in the early stages of primary progressive multiple sclerosis (PPMS), affecting both grey and white matter (GM and WM). However, to date no clinical or radiological predictors of GM and WM atrophy have been identified. The aim was to investigate short-term changes in GM and WM volumes and to assess the predictive value of demographic, clinical and radiological variables in order to gain a better understanding of the pathological substrate underlying these changes. Thirty-one subjects with PPMS within 5 years of symptom onset were studied at baseline and after 1 year. At baseline, patients underwent neurological examination and were scored on the Expanded Disability Status Scale (EDSS) and Multiple Sclerosis Functional Composite. They had 3D inversion-prepared fast spoiled gradient recalled (FSPGR), dual-echo and triple-dose post-contrast T1-weighted spin echo MRI scans. Proton density and enhancing lesion loads were determined. The 3DFSPGR sequence was repeated after 1 year and brain volume changes were calculated using two techniques, SPM99 (statistical parametric mapping) and SIENA (structural image evaluation, using normalization, of atrophy). Stepwise linear regression models were applied to baseline variables to identify independent predictors of atrophy development. Using SPM99, a decrease in brain parenchymal fraction (−1.03%; P < 0.001) and GM fraction (−1.49%; P < 0.001) was observed. The number of enhancing lesions independently predicted decrease in brain parenchymal fraction (P = 0.019) and decrease in WM fraction (P = 0.002). No independent predictors of GM fraction decrease were found. A mean brain volume change of −0.63% (range −4.27% to +1.18%; P = 0.002) was observed using SIENA, which was independently predicted by EDSS (P = 0.004). Global and GM atrophy can be detected over a 1-year period in early PPMS. The former may be predicted by the degree of inflammation, while the latter seems to be independent of it. SIENA and SPM-based methods appear to provide complementary information.
Introduction
Atrophy is a relatively well-established marker of axonal loss and disability in multiple sclerosis (Losseff et al., 1996; Lycklama à Nijeholt et al., 1998; Rovaris et al., 2001; Miller et al., 2002). However, it has been argued that the value of atrophy as a marker is limited by the fact that it represents the final step in the chain of pathological changes leading to tissue loss, and, as a consequence, disability changes related to brain atrophy are likely to be irrecoverable (Miller et al., 2002). However, the ability to predict atrophy development is of clinical relevance, as it will help predict irreversible disability. Until recently, atrophy measures were based on partial (Losseff et al., 1996), indirect (Lycklama à Nijeholt et al., 1998) or global (Rudick et al., 1999) techniques unable to focus on specific brain tissue types. The development of segmentation-based methods (Miller et al., 2002) has allowed white and grey matter volumes to be assessed separately. Even though multiple sclerosis has long been considered a white matter condition, the involvement of grey matter in its pathology (Kidd et al., 1999; Peterson et al., 2001) and its relevance to disability has recently been reinforced (De Stefano et al., 2003), highlighting a potential role for tissue-specific measures of atrophy.
Primary progressive multiple sclerosis (PPMS) is a useful model to investigate the evolution of atrophy and its predictors, as the potential confounders of inflammation and oedema play a lesser role (Thompson et al., 1991; Revesz et al., 1994). However, findings in PPMS must be generalized with caution, as the pathological processes leading to atrophy might vary in different forms of the condition. The present study focuses on patients with early PPMS, as they are more likely to display a closer relationship between clinical and radiological variables than those with more established disease (Cottrell et al., 1999; Sastre-Garriga et al., 2004). We used a method based on SPM segmentation (statistical parametric mapping; Functional Imaging Laboratory, University College London) to obtain quantitative information on grey and white matter volume. Recognizing that different atrophy measurement methods have different potential for bias, the MRI data have also been processed using another fully automated and well tested methodology to assess the evolution of whole brain atrophy over time: SIENA (structural image evaluation, using normalization, of atrophy; Centre for Functional Magnetic Resonance Imaging of the Brain, Oxford) (Smith et al., 2002).
Thus, this work set out to study the evolution of tissue-specific brain atrophy in patients with clinically early PPMS. Its applications are twofold: first, to generate a hypothesis on the pathological substrate underlining the changes observed, and secondly to identify factors which may predict these changes and to use these changes themselves to evaluate interventions.
Methods
Patients
Thirty-one patients with PPMS within 5 years of disease onset were recruited at the National Hospital for Neurology and Neurosurgery, Queen Square, London. All patients fulfilled the PPMS diagnostic criteria (definite or probable) (Thompson et al., 2000). These patients are part of a larger cohort of patients with clinically early PPMS from which baseline results are already available (Sastre-Garriga et al., 2004). All patients underwent conventional neurological assessment and were scored on the Expanded Disability Status Scale (EDSS) and Multiple Sclerosis Functional Composite (MSFC) at baseline. Z scores for the MSFC subtests were calculated using the published methods with our own sample as a reference (Cutter et al., 1999). One patient had a course of oral steroids 3 months before the second scan, and no patient was on disease-modifying drugs. The study was approved by the joint ethics committee of the National Hospital for Neurology and Neurosurgery and the Institute of Neurology (London) prior to study initiation. All subjects gave written informed consent before being included in the study.
MRI methods
Scan acquisition
All images were acquired using a 1.5 T Signa scanner (General Electric, Milwaukee, WI, USA). No major hardware upgrades were carried out on the scanner, and weekly quality assurance sessions confirmed the stability of measurements throughout the study. Three sets of images were acquired at baseline: a 3D inversion-prepared fast spoiled gradient recalled (3DFSPGR) sequence [124 contiguous axial slices, echo time (TE) 4.2 ms, repetition time (TR) 13.3 ms, inversion time 450 ms, number of excitations (NEX) 1, field of view (FOV) 300 × 225 mm over an image matrix of 256 × 160 (interpolated to a 256 × 256 reconstructed matrix for a final x–y in-plane resolution of 1.17 × 1.17 mm), slice thickness (ST) 1.5 mm]; a dual spin echo sequence (28 contiguous slices, TE 30/80 ms, TR 1720 ms, NEX 0.75, ST 5 mm); and a T1-weighted spin echo sequence (28 contiguous axial slices, TE 20 ms, TR 540 ms, NEX 1, flip angle 90°, ST 5 mm), acquired before and 5 min after intravenous administration of triple-dose gadolinium (0.6 ml/kg of body weight; Magnevist, Schering, Germany). For the sake of patient comfort, scan sessions were split into two, and T1 gadolinium-enhanced scans were acquired in a separate session; the mean separation between sessions was 14.8 days (median 7; interquartile range 7–24). The 3DFSPGR sequence was acquired again 1 year later (mean time between acquisitions 12.2 months; interquartile range 11.4–13.1).
Image analysis
Lesion loads were estimated from proton density-weighted images and gadolinium-enhanced baseline scans using Dispimage (D. L. Plummer, University College London, London, UK). 3DFSPGR images from baseline and 1-year scans were segmented into white matter (WM), grey matter (GM) and cerebrospinal fluid (CSF) using SPM99 (available at http://www.fil.ion.ucl.ac.uk/spm/) following a previously described method (Chard et al., 2002a; Sastre-Garriga et al., 2004). Using in-house software, lesion masks were obtained by contouring lesion on the 3DFSPGR scans; these masks were subtracted from WM, GM and CSF segmentations to obtain four mutually exclusive tissue masks with their volumes in millilitres. Total intracranial volume (TIV) was calculated as WM + GM + CSF + lesion mask volumes. Brain parenchymal fraction was calculated as (WM + GM + lesion mask volumes)/TIV, white matter fraction as (WM + lesion mask volumes)/TIV, and grey matter fraction as GM volume/TIV. Brain atrophy was also estimated on 3DFSPGR images from baseline and 1-year scans using SIENA version 2.2 (available at http://www.fmrib.ox.ac.uk/analysis/research/siena/). Briefly, SIENA is a fully automated method of analysis which works on serially acquired images and performs brain extraction on both baseline and follow-up images, followed by registration of both brain images to obtain an estimate of the percentage brain volume change (PBVC) between scans (Smith et al., 2002). Coefficients of variation for SPM are as follows: 0.2% for brain parenchymal fraction, 0.4% for grey matter fraction and 0.9% for white matter fraction (Chard et al., 2002a). For PBVC the median absolute error is 0.15% (Smith et al., 2002).
Statistical methods
Statistical analysis was performed using SPSS 11.5 (SPSS, Chicago, IL, USA). A paired samples t-test was performed to detect statistically significant differences between baseline and 1-year estimates of atrophy (brain parenchymal, grey and white matter fractions) and a one sample t-test for PBVC. Percentage changes in tissue fractions were obtained by subtracting the baseline from the 1-year follow-up estimates and dividing by the baseline values, then multiplying by 100. Pearson correlation coefficients were used to assess the presence of linear associations between PBVC, the percentage change in tissue fractions (brain parenchymal, grey and white matter fractions) and clinical and radiological variables, except for EDSS, when Spearman rank correlation coefficients were calculated. Significance values for correlation coefficients are reported without correction for multiple comparisons to avoid type II errors (Perneger, 1998). Stepwise linear regression was performed with PBVC and the per centage change in tissue fractions (brain parenchymal, grey and white matter fractions) as dependent variables against three sets of independent variables: clinical (age, disease duration, EDSS, MSFC), radiological (age, disease duration, number of gadolinium-enhancing lesions, proton density lesion volume) and combined (age, disease duration, EDSS, MSFC, number of gadolinium-enhancing lesions, proton density lesion volume).
Results
Sample characteristics (Table 1)
The 31 patients with clinically early PPMS (less than 5 years of disease duration) had a mean follow-up time of 12.2 months (interquartile range 11.4–13.1). There were 18 males and 13 females; median age at baseline was 46.0 years (range 26–62) and median disease duration was 3.0 (range 2–5). Median EDSS at baseline was 4.5 (range 3.5 range: 7.0) and mean MSFC was −0.3162 (SD 1.44). Mean proton density lesion load was 26.01 ml (SD 22.97) and the mean number of gadolinium-enhancing lesions was 0.96 (SD 1.44).
Baseline clinical, demographic and radiological features of patients
| Patients (n = 31) | |
|---|---|
| Sex ratio (male/female) | 58%/42% |
| Age at scanning* | 46 (26–62) |
| Disease duration (years)* | 3.0 (2–5) |
| EDSS* | 4.5 (3.5–7) |
| MSFC** | −0.316 (1.44) |
| Brain parenchymal fraction** | 0.796 (0.035) |
| White matter fraction** | 0.266 (0.022) |
| Grey matter fraction** | 0.529 (0.023) |
| Number of gadolinium-positive lesions** | 0.96 (1.44) |
| Patients with gadolinium-positive lesions | 48.4% |
| Proton density lesion volume**,*** | 26.01 (22.97) |
| Patients (n = 31) | |
|---|---|
| Sex ratio (male/female) | 58%/42% |
| Age at scanning* | 46 (26–62) |
| Disease duration (years)* | 3.0 (2–5) |
| EDSS* | 4.5 (3.5–7) |
| MSFC** | −0.316 (1.44) |
| Brain parenchymal fraction** | 0.796 (0.035) |
| White matter fraction** | 0.266 (0.022) |
| Grey matter fraction** | 0.529 (0.023) |
| Number of gadolinium-positive lesions** | 0.96 (1.44) |
| Patients with gadolinium-positive lesions | 48.4% |
| Proton density lesion volume**,*** | 26.01 (22.97) |
Median (range);
mean (SD);
Lesion volume in ml;
EDSS = Expanded Disability Status Scale; MSFC = Multiple Sclerosis Functional Composite.
Baseline clinical, demographic and radiological features of patients
| Patients (n = 31) | |
|---|---|
| Sex ratio (male/female) | 58%/42% |
| Age at scanning* | 46 (26–62) |
| Disease duration (years)* | 3.0 (2–5) |
| EDSS* | 4.5 (3.5–7) |
| MSFC** | −0.316 (1.44) |
| Brain parenchymal fraction** | 0.796 (0.035) |
| White matter fraction** | 0.266 (0.022) |
| Grey matter fraction** | 0.529 (0.023) |
| Number of gadolinium-positive lesions** | 0.96 (1.44) |
| Patients with gadolinium-positive lesions | 48.4% |
| Proton density lesion volume**,*** | 26.01 (22.97) |
| Patients (n = 31) | |
|---|---|
| Sex ratio (male/female) | 58%/42% |
| Age at scanning* | 46 (26–62) |
| Disease duration (years)* | 3.0 (2–5) |
| EDSS* | 4.5 (3.5–7) |
| MSFC** | −0.316 (1.44) |
| Brain parenchymal fraction** | 0.796 (0.035) |
| White matter fraction** | 0.266 (0.022) |
| Grey matter fraction** | 0.529 (0.023) |
| Number of gadolinium-positive lesions** | 0.96 (1.44) |
| Patients with gadolinium-positive lesions | 48.4% |
| Proton density lesion volume**,*** | 26.01 (22.97) |
Median (range);
mean (SD);
Lesion volume in ml;
EDSS = Expanded Disability Status Scale; MSFC = Multiple Sclerosis Functional Composite.
Evolution of brain atrophy (Table 2, Fig. 1)
Using the SPM-based segmentation method, significant decreases (P < 0.001) were observed in both brain parenchymal fraction (mean −1.03%; SD 1.3%) and grey matter fraction (mean −1.50%; SD 1.6), whereas white matter fraction did not change significantly (−0.07%; P = 0.749). Using SIENAv2.2 a decrease of −0.63% (SD 1.05) was observed (P = 0.002).
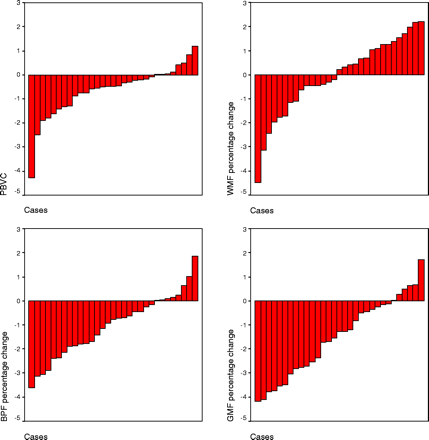
Bar graphs displaying percentage brain volume change (PBVC) obtained with SIENAv2.2 (top left) and percentage changes in brain parenchymal fraction (BPF; bottom left), white matter fraction (WMF; top right) and grey matter fraction (GMF; bottom right) obtained with SPM-based methods.
Values for brain parenchymal fraction, white matter fraction and grey matter fraction obtained with SPM-based methods and for PBVC obtained with SIENAv2.2
| Baseline | 12 months | P | |
|---|---|---|---|
| Brain parenchymal fraction* | 0.796 (0.035) | 0.787 (0.038) | <0.001 |
| White matter fraction* | 0.266 (0.022) | 0.266 (0.022) | 0.749 |
| Grey matter fraction* | 0.529 (0.023) | 0.521 (0.025) | <0.001 |
| PBVC*,** | – | −0.636 (1.05) | 0.002 |
| Baseline | 12 months | P | |
|---|---|---|---|
| Brain parenchymal fraction* | 0.796 (0.035) | 0.787 (0.038) | <0.001 |
| White matter fraction* | 0.266 (0.022) | 0.266 (0.022) | 0.749 |
| Grey matter fraction* | 0.529 (0.023) | 0.521 (0.025) | <0.001 |
| PBVC*,** | – | −0.636 (1.05) | 0.002 |
Mean (standard deviation);
Values for PBVC represent percentage changes from baseline examination;
PVBC = percentage brain volume change.
Values for brain parenchymal fraction, white matter fraction and grey matter fraction obtained with SPM-based methods and for PBVC obtained with SIENAv2.2
| Baseline | 12 months | P | |
|---|---|---|---|
| Brain parenchymal fraction* | 0.796 (0.035) | 0.787 (0.038) | <0.001 |
| White matter fraction* | 0.266 (0.022) | 0.266 (0.022) | 0.749 |
| Grey matter fraction* | 0.529 (0.023) | 0.521 (0.025) | <0.001 |
| PBVC*,** | – | −0.636 (1.05) | 0.002 |
| Baseline | 12 months | P | |
|---|---|---|---|
| Brain parenchymal fraction* | 0.796 (0.035) | 0.787 (0.038) | <0.001 |
| White matter fraction* | 0.266 (0.022) | 0.266 (0.022) | 0.749 |
| Grey matter fraction* | 0.529 (0.023) | 0.521 (0.025) | <0.001 |
| PBVC*,** | – | −0.636 (1.05) | 0.002 |
Mean (standard deviation);
Values for PBVC represent percentage changes from baseline examination;
PVBC = percentage brain volume change.
Brain atrophy predictors: univariate analysis (Table 3)
No significant gender differences were found in the development of atrophy for any of the measures studied. Significant linear associations were found between enhancing lesion numbers at baseline and PVBC (r = −0.458, P = 0.010), brain parenchymal fraction percentage change (r = −0.419, P = 0.019) and white matter fraction percentage change (r = −0.532, P = 0.002). Brain parenchymal fraction percentage change was also associated with baseline proton density lesion load (r = −0.364, P = 0.044) and a significant linear association was found between EDSS at baseline and PVBC over the 12 months of the study (r = −0.509, P = 0.003). No significant associations for grey matter fraction change were found with any of the variables tested.
Correlations of PBVC, brain parenchymal fraction, white matter fraction and grey matter fraction percentage changes after 12 months with demographic, clinical and radiological measures at baseline
| r | P | |||
|---|---|---|---|---|
| PBVC | ||||
| Age | 0.107 | 0.567 | ||
| Disease duration | 0.136 | 0.466 | ||
| EDSS* | −0.509 | 0.003 | ||
| MSFC | 0.190 | 0.306 | ||
| Proton density lesion load | −0.202 | 0.275 | ||
| Enhancing lesion numbers | −0.458 | 0.010 | ||
| Brain parenchymal fraction (% change) | ||||
| Age | 0.286 | 0.119 | ||
| Disease duration | 0.288 | 0.117 | ||
| EDSS* | −0.165 | 0.375 | ||
| MSFC | 0.056 | 0.763 | ||
| Proton density lesion load | −0.364 | 0.044 | ||
| Enhancing lesion numbers | −0.419 | 0.019 | ||
| White matter fraction (% change) | ||||
| Age | 0.304 | 0.096 | ||
| Disease duration | 0.325 | 0.074 | ||
| EDSS* | −0.154 | 0.407 | ||
| MSFC | 0.160 | 0.390 | ||
| Proton density lesion load | −0.177 | 0.342 | ||
| Enhancing lesion numbers | −0.532 | 0.002 | ||
| Grey matter fraction (% change) | ||||
| Age | −0.174 | 0.349 | ||
| Disease duration | 0.185 | 0.319 | ||
| EDSS* | −0.079 | 0.673 | ||
| MSFC | −0.012 | 0.950 | ||
| Proton density lesion load | −0.341 | 0.060 | ||
| Enhancing lesion numbers | −0.237 | 0.199 | ||
| r | P | |||
|---|---|---|---|---|
| PBVC | ||||
| Age | 0.107 | 0.567 | ||
| Disease duration | 0.136 | 0.466 | ||
| EDSS* | −0.509 | 0.003 | ||
| MSFC | 0.190 | 0.306 | ||
| Proton density lesion load | −0.202 | 0.275 | ||
| Enhancing lesion numbers | −0.458 | 0.010 | ||
| Brain parenchymal fraction (% change) | ||||
| Age | 0.286 | 0.119 | ||
| Disease duration | 0.288 | 0.117 | ||
| EDSS* | −0.165 | 0.375 | ||
| MSFC | 0.056 | 0.763 | ||
| Proton density lesion load | −0.364 | 0.044 | ||
| Enhancing lesion numbers | −0.419 | 0.019 | ||
| White matter fraction (% change) | ||||
| Age | 0.304 | 0.096 | ||
| Disease duration | 0.325 | 0.074 | ||
| EDSS* | −0.154 | 0.407 | ||
| MSFC | 0.160 | 0.390 | ||
| Proton density lesion load | −0.177 | 0.342 | ||
| Enhancing lesion numbers | −0.532 | 0.002 | ||
| Grey matter fraction (% change) | ||||
| Age | −0.174 | 0.349 | ||
| Disease duration | 0.185 | 0.319 | ||
| EDSS* | −0.079 | 0.673 | ||
| MSFC | −0.012 | 0.950 | ||
| Proton density lesion load | −0.341 | 0.060 | ||
| Enhancing lesion numbers | −0.237 | 0.199 | ||
All correlation coefficients reported are Pearson correlation coefficients except for EDSS (Spearman);
Significant correlations appear in bold. PBVC = percentage brain volume change obtained with SIENAv2.2.
Correlations of PBVC, brain parenchymal fraction, white matter fraction and grey matter fraction percentage changes after 12 months with demographic, clinical and radiological measures at baseline
| r | P | |||
|---|---|---|---|---|
| PBVC | ||||
| Age | 0.107 | 0.567 | ||
| Disease duration | 0.136 | 0.466 | ||
| EDSS* | −0.509 | 0.003 | ||
| MSFC | 0.190 | 0.306 | ||
| Proton density lesion load | −0.202 | 0.275 | ||
| Enhancing lesion numbers | −0.458 | 0.010 | ||
| Brain parenchymal fraction (% change) | ||||
| Age | 0.286 | 0.119 | ||
| Disease duration | 0.288 | 0.117 | ||
| EDSS* | −0.165 | 0.375 | ||
| MSFC | 0.056 | 0.763 | ||
| Proton density lesion load | −0.364 | 0.044 | ||
| Enhancing lesion numbers | −0.419 | 0.019 | ||
| White matter fraction (% change) | ||||
| Age | 0.304 | 0.096 | ||
| Disease duration | 0.325 | 0.074 | ||
| EDSS* | −0.154 | 0.407 | ||
| MSFC | 0.160 | 0.390 | ||
| Proton density lesion load | −0.177 | 0.342 | ||
| Enhancing lesion numbers | −0.532 | 0.002 | ||
| Grey matter fraction (% change) | ||||
| Age | −0.174 | 0.349 | ||
| Disease duration | 0.185 | 0.319 | ||
| EDSS* | −0.079 | 0.673 | ||
| MSFC | −0.012 | 0.950 | ||
| Proton density lesion load | −0.341 | 0.060 | ||
| Enhancing lesion numbers | −0.237 | 0.199 | ||
| r | P | |||
|---|---|---|---|---|
| PBVC | ||||
| Age | 0.107 | 0.567 | ||
| Disease duration | 0.136 | 0.466 | ||
| EDSS* | −0.509 | 0.003 | ||
| MSFC | 0.190 | 0.306 | ||
| Proton density lesion load | −0.202 | 0.275 | ||
| Enhancing lesion numbers | −0.458 | 0.010 | ||
| Brain parenchymal fraction (% change) | ||||
| Age | 0.286 | 0.119 | ||
| Disease duration | 0.288 | 0.117 | ||
| EDSS* | −0.165 | 0.375 | ||
| MSFC | 0.056 | 0.763 | ||
| Proton density lesion load | −0.364 | 0.044 | ||
| Enhancing lesion numbers | −0.419 | 0.019 | ||
| White matter fraction (% change) | ||||
| Age | 0.304 | 0.096 | ||
| Disease duration | 0.325 | 0.074 | ||
| EDSS* | −0.154 | 0.407 | ||
| MSFC | 0.160 | 0.390 | ||
| Proton density lesion load | −0.177 | 0.342 | ||
| Enhancing lesion numbers | −0.532 | 0.002 | ||
| Grey matter fraction (% change) | ||||
| Age | −0.174 | 0.349 | ||
| Disease duration | 0.185 | 0.319 | ||
| EDSS* | −0.079 | 0.673 | ||
| MSFC | −0.012 | 0.950 | ||
| Proton density lesion load | −0.341 | 0.060 | ||
| Enhancing lesion numbers | −0.237 | 0.199 | ||
All correlation coefficients reported are Pearson correlation coefficients except for EDSS (Spearman);
Significant correlations appear in bold. PBVC = percentage brain volume change obtained with SIENAv2.2.
Brain atrophy predictors: stepwise regression models; dependent variables
Brain parenchymal fraction percentage change over 12 months
No independent predictor was found in the clinical model; the number of enhancing lesions was the only independent predictor of brain parenchymal fraction percentage change in both the radiological and combined models (R2 = 0.17, P = 0.019).
PBVC over 12 months
EDSS was found to be an independent predictor when applied to the clinical model (R2 = 0.25, P = 0.004); on application of the radiological model the number of enhancing lesions was found to be the only independent predictor (R2 = 0.21, P = 0.010). Only EDSS at baseline was found to be independently associated (R2 = 0.25, P = 0.004) in the combined model.
White matter fraction percentage change over 12 months
No independent predictor was found in the clinical model; the number of enhancing lesions was found to be the only independent predictor of white matter fraction percentage change (R2 = 0.28; P = 0.002) in the radiological model; on applying the combined model a similar result was obtained, with the number of enhancing lesions as the only independent predictor (R2 = 0.28; P = 0.002).
Grey matter fraction percentage change over 12 months
No significant predictors were found in any of the three models.
Discussion
The findings of the present study may be summarized as: (i) changes in grey but not white matter volume can be detected in patients with clinically early PPMS over periods of time as brief as 1 year; (ii) the number of enhancing lesions at baseline is predictive of white matter atrophy development; (iii) grey matter volume loss cannot be predicted using conventional clinical and MRI parameters; and (iv) different methodological approaches to the estimation of atrophy may provide complementary information which is relevant to the clinical and radiological associations found.
Volume changes over 1 year
Changes in atrophy have been detected in patients with established PPMS over 1 year using non-normalized partial brain measures (Stevenson et al., 2000). This has also been shown, using longer follow-up times, for normalized global brain measures (Kalkers et al., 2002). The present study suggests that, over the short term, the development of atrophy in patients with early PPMS is due mainly to change in grey matter. It is also worth noting that the observed decrease in grey matter fraction seems to be a real finding and not a methodological artefact, as it occurs in spite of the bias in SPM segmentation, whereby in subjects with increasing lesion loads SPM tends to overestimate grey matter volumes (Chard et al., 2002a). A possible explanation for the findings of this study would be that inflammation and gliosis may be obscuring white matter volume loss, whereas grey matter tissue loss occurs without reactive gliosis or inflammation that may mask volume loss (Peterson et al., 2001). The present findings are also consistent with two longitudinal studies from our group, which have assessed the evolution of grey and white matter atrophy in patients with clinically isolated syndromes (Dalton et al., 2004) and relapsing–remitting multiple sclerosis (Chard et al., 2004). Both studies have found a significant decrease in grey matter fraction with no concomitant decrease in white matter fraction, although white matter fraction was reduced at baseline in early relapsing–remitting and PPMS when compared with healthy control subjects, suggesting that it may be an early feature (Chard et al., 2002b; Sastre-Garriga et al., 2004). As patients from both studies are in the early clinical stages of the condition this might indicate that white matter loss has already taken place in a preclinical stage.
The number of enhancing lesions predicts the development of white matter atrophy
Many studies have assessed the effect of enhancing lesions on subsequent brain volume changes in relapsing–remitting multiple sclerosis (Rudick et al., 1999; Saindane et al., 2000; Fisher et al., 2002; Hardmeier et al., 2003), clinically isolated syndromes suggestive of multiple sclerosis (Dalton et al., 2004; Paolillo et al., 2004) and secondary progressive multiple sclerosis (Inglese et al., 2004). Most have found that large volumes of enhancing lesions are predictive of or associated with the development of atrophy. We are not aware of any similar studies in patients with PPMS. Resolution of inflammation-associated oedema is likely to be an important contributing factor in white matter volume changes, but the contribution of inflammation to subsequent white matter tissue loss cannot be ruled out. Ensuing gliosis, which might prevent volume loss, might complicate the picture even further. In this regard, Inglese and colleagues have shown, in a group of patients with rapidly evolving secondary progressive multiple sclerosis and a percentage of annual global tissue loss of −1.88%, that even after suppression of inflammation through therapy with autologous haematopoietic stem cell transplantation, subsequent brain atrophy development is still marginally associated with the enhancing lesion load prior to treatment (Inglese et al., 2004).
Failure to predict grey matter atrophy
The baseline, cross-sectional analysis of this cohort has shown that grey matter fraction is not associated with white matter lesion loads (T2 and gadolinium-enhancing). This is in contrast with a previous study by our own group, in early relapsing–remitting multiple sclerosis, in which correlations were found between white matter lesion load and grey matter fraction that were thought to reflect dying back and Wallerian degeneration processes (Chard et al., 2002b). The phenotypic difference between the cohorts studied (early relapsing–remitting multiple sclerosis versus early PPMS) may help explain this difference. Moreover, the possible relationship between lesions in white matter and grey matter atrophy may be affected by the presence of grey matter lesions, which are difficult to observe using conventional MRI but are known to be numerous (Bö et al., 2003), although no data are available comparing the extent and severity of grey matter lesions in PPMS and relapsing–remitting multiple sclerosis. Given that previous studies have demonstrated a relationship between cortical grey matter atrophy and functional activity and clinical disability (De Stefano et al., 2003; Filippi and Rocca, 2003), the identification of predictors of grey matter loss or dysfunction is an important focus of future research. Its independence from gross inflammation may make the measurement of grey matter volume changes a useful surrogate for the assessment of new neuroprotective strategies in multiple sclerosis.
Role of methodology
The estimates of percentage global tissue loss in this study, −1.03% for SPM-based methodology and −0.63% for SIENAv2.2, are within the range seen in different phenotypes of multiple sclerosis, which goes from −0.46 to −1.9% (Inglese et al., 1999; Dalton et al., 2004), and uses a range of atrophy techniques. Although separate grey and white matter volumes cannot be obtained, SIENA is more user-friendly as it needs no manual operator input and the software is freely available on the World-Wide Web in a compact package. Conversely, SPM-derived methods need considerable operator input (lesion contouring), and, although it can also be downloaded from the World-Wide Web, in-house software is required to generate masks and final volume outputs. Therefore, it could be suggested that SPM-based methods be used for research purposes and SIENA v2.2 in clinical applications. However, even though the results of the present study are in keeping with other studies using SPM (Dalton et al., 2004) and SIENA (Jasperse et al., 2004), it is important to note that markedly different associations between atrophy development and baseline variables have been observed using the two methods. Estimates of atrophy are known to differ depending on the segmentation procedure and pulse sequence used for their estimation (Leigh et al., 2002; Horsfield et al., 2003). SPM segmentation is biased by the presence of lesions, although this bias seems to have an effect on grey and white matter only and not whole-brain estimates (Chard et al., 2002a). A trend for underestimation of higher atrophy has been reported with SIENA (Smith et al., 2002). More methodological studies are warranted before the present findings can be fully explained.
In summary, progressive grey matter atrophy can be detected in early PPMS over 1-year periods but cannot be predicted by conventional clinical and MRI variables, while white matter volume changes are influenced by acute inflammation even in PPMS.
We wish to thank the subjects who kindly agreed to take part in this study. We acknowledge the support of the Wellcome Trust (G.T.I.), the Spanish Ministry of Health (J.S.G., BEFI 02/9115) and the Multiple Sclerosis Society of Great Britain and Northern Ireland.
References
Bö L, Vedeler CA, Nyland HI, Trapp BD, Mörk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients.
Chard DT, Parker GJM, Griffin CMB, Thompson AJ, Miller DH. The reproducibility and sensitivity of brain tissue volume measurements derived from an SPM-based segmentation methodology.
Chard DT, Griffin CM, Parker GJM, Kapoor R, Thompson AJ, Miller DH. Brain atrophy in clinically early relapsing-remitting multiple sclerosis.
Chard DT, Griffin CM, Rashid W, Davies GR, Altmann DR, Kapoor R, et al. Progressive grey matter atrophy in clinically early relapsing-remitting multiple sclerosis.
Cottrell DA, Kremenchutzky M, Rice GPA, Koopman WJ, Hader W, Baskerville J, et al. The natural history of multiple sclerosis: a geographically based study. 5. The clinical features and natural history of primary progressive multiple sclerosis.
Cutter GR, Baier ML, Rudick RA, Cookfair DL, Fischer JS, Petkau J, et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure.
Dalton CM, Chard DT, Davies GR, Myszkiel KA, Altmann DR, Fernando K, et al. Early development of multiple sclerosis is associated with progressive grey matter atrophy in patients presenting with clinically isolated syndromes.
De Stefano N, Matthews PM, Filippi M, Agosta F, De Luca M, Bartolozzi ML, et al. Evidence of early cortical atrophy in MS. Relevance to white matter changes and disability.
Filippi M, Rocca MA. Disturbed function and plasticity in multiple sclerosis as gleaned from functional magnetic resonance imaging.
Fisher E, Rudick RA, Simon JH, Cutter G, Baier M, Lee JC, et al. Eight-year follow-up study of brain atrophy in patients with MS.
Hardmeier M, Wagenpfeil S, Freitag P, Fisher E, Rudick RA, Kooijmans-Coutinho M, et al. Atrophy is detectable within a 3-month period in untreated patients with active relapsing remitting multiple sclerosis.
Horsfield MA, Rovaris M, Rocca MA, Rossi P, Benedict RH, Filippi M, et al. Whole-brain atrophy in multiple sclerosis measured by two segmentation processes from various MRI sequences.
Inglese M, Rovaris M, Giacomotti L, Mastronardo G, Comi G, Filippi M. Quantitative brain volumetric analysis from patients with multiple sclerosis: a follow-up study.
Inglese M, Mancardi GL, Pagani E, Rocca MA, Murialdo A, Saccardi R, et al. Brain tissue loss occurs after suppression of enhancement in patients with multiple sclerosis treated with autologous haematopoietic stem cell transplantation.
Jasperse MMS, Minneboo A, Kalkers NF, De Groot V, Karas GB, Uitdehaag BMJ, et al. Brain volume measurement in early MS: clinical/radiological correlates and predictors.
Kalkers NF, Ameziane N, Bot JCJ, Minneboo A, Polman CH, Barkhof F. Longitudinal brain volume measurement in multiple sclerosis.
Kidd D, Barkhof F, McConnell R, Algra PR, Allen IV, Revesz T. Cortical lesions in multiple sclerosis.
Leigh R, Ostuni J, Pham D, Goldszal A, Lewis BK, Howard T, et al. Estimating cerebral atrophy in multiple sclerosis patients from various MR pulse sequences.
Lycklama à Nijeholt GJ, van Walderveen MA, Castelijns JA, van Waesberghe JH, Polman C, Scheltens P, et al. Brain and spinal cord abnormalities in multiple sclerosis. Correlation between MRI parameters, clinical subtypes and symptoms.
Losseff NA, Wang L, Lai HM, Yoo DS, Gawne-Cain ML, McDonald WI, et al. Progressive cerebral atrophy in multiple sclerosis.
Miller DH, Barkhof F, Frank JA, Parker GJM, Thompson AJ. Measurement of atrophy in multiple sclerosis: pathological basis, methodological aspects and clinical relevance.
Paolillo A, Piattella MC, Pantano P, Di Legge S, Caramia F, Russo P, et al. The relationship between inflammation and atrophy in clinically isolated syndromes suggestive of multiple sclerosis. A monthly MRI study after triple-dose gadolinium-DTPA.
Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions.
Revesz T, Kidd D, Thompson AJ, Barnard RO, McDonald WI. A comparison of the pathology of primary and secondary progressive multiple sclerosis.
Rovaris M, Comi G, Rocca MA, Wolinsky JS, Filippi M. Short-term brain volume changes in relapsing-remitting multiple sclerosis. Effect of glatiramer acetate and implications.
Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L. Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS.
Saindane AM, Ge Y, Udupa JK, Babb JS, Mannon LJ, Grossman RI. The effect of gadolinium-enhancing lesions on whole brain atrophy in relapsing-remitting MS.
Sastre-Garriga J, Ingle GT, Chard DT, Ramió-Torrentà Ll, Miller DH, Thompson AJ. Grey and white matter atrophy in early clinical stages of primary progressive multiple sclerosis.
Smith SM, Zhang Y, Jenkinson M, Chen J, Matthews PM, Federico A, et al. Accurate, robust, and automated longitudinal and cross-sectional brain change analysis.
Stevenson VL, Miller DH, Leary SM, Rovaris M, Barkhof F, Brochet B, et al. One year follow up study of primary and transitional progressive multiple sclerosis.
Thompson AJ, Kermode AG, Wicks D, MacManus DG, Kendall BE, Kingsley DPE, et al. Major differences in the dynamics of primary and secondary progressive multiple sclerosis.


{kind=link}