



Abstract
Infantile onset spinocerebellar ataxia (IOSCA) (MIM 271245) is a severe autosomal recessively inherited neurodegenerative disorder characterized by progressive atrophy of the cerebellum, brain stem and spinal cord and sensory axonal neuropathy. We report here the molecular background of this disease based on the positional cloning/candidate approach of the defective gene. Having established the linkage to chromosome 10q24, we restricted the critical DNA region using single nucleotide polymorphism-based haplotypes. After analyzing all positional candidate transcripts, we identified two point mutations in the gene C10orf2 encoding Twinkle, a mitochondrial deoxyribonucleic acid (mtDNA)-specific helicase, and a rarer splice variant Twinky, underlying IOSCA. The founder IOSCA mutation, homozygous in all but one of the patients, leads to a Y508C amino acid change in the polypeptides. One patient, heterozygous for Y508C, carries a silent coding region cytosine to thymine transition mutation in his paternal disease chromosome. This allele is expressed at a reduced level, causing the preponderance of messenger RNAs encoding Y508C polypeptides and thus leads to the IOSCA disease phenotype. Previously, we have shown that different mutations in this same gene cause autosomal dominant progressive external ophthalmoplegia (adPEO) with multiple mtDNA deletions (MIM 606075), a neuromuscular disorder sharing a spectrum of symptoms with IOSCA. IOSCA phenotype is the first recessive one due to Twinkle and Twinky mutations, the dominant PEO mutations affecting mtDNA maintenance, but in IOSCA, mtDNA stays intact. The severe neurological phenotype observed in IOSCA, a result of only a single amino acid substitution in Twinkle and Twinky, suggests that these proteins play a crucial role in the maintenance and/or function of specific affected neuronal subpopulations.
INTRODUCTION
Infantile onset spinocerebellar ataxia (IOSCA) (MIM 271245) is a severe autosomal recessively inherited neurodegenerative disorder of originally unknown cause. It manifests at the age of 9–18 months in previously healthy infants as ataxia, athetosis, muscle hypotonia and loss of deep tendon reflexes, and at the later stage, as hypacusis, ophthalmoplegia, optic atrophy and female primary hypogonadism of the hypergonadotropic type. The cause of premature death has often been prolonged epileptic seizures. Morphologically, IOSCA is characterized by sensory axonal neuropathy and progressive atrophy of the cerebellum, brain stem and spinal cord as prominent morphological features (1–3). A most typical representative of the Finnish disease heritage (4,5), IOSCA has so far been described in 21 Finnish patients in 15 nuclear families (1,6). Nosologically, IOSCA has been classified to belong to the heterogeneous group of hereditary ataxia syndromes (1). These are characterized by various signs and symptoms originating mainly from the degeneration of the cerebellum, brain stem and spinal cord and can show either autosomal recessive or autosomal dominant inheritance (7). Among the ataxic syndromes, IOSCA bears both neuropathologically and phenotypically closest resemblance to the most common recessively inherited ataxia, that of Friedreich's ataxia (FRDA) (MIM 606829), known to be caused by the lack of frataxin, a nuclear DNA-encoded mitochondrial protein affecting cellular iron homeostasis (reviewed in 8). However, the cardiac involvement of FRDA is not a feature in IOSCA, nor has mitochondrial dysfunction been observed in the IOSCA patients. We previously mapped the IOSCA locus to chromosome 10q24 and thereafter restricted the critical region by haplotype analyses. All but one of the IOSCA patients were shown to carry a common core haplotype in homozygous form, indicating a single old founder mutation (9,10). One compound heterozygote patient had a totally different haplotype in his paternal disease chromosome, implying the presence of a second minor mutation (6). Here we report the refinement of the IOSCA locus, analyses of the positional candidate genes and the identification of the defective gene by pinpointing two mutations underlying IOSCA.
RESULTS
Single nucleotide polymorphism (SNP) haplotypes and restriction of the area
By studying extended SNP haplotypes of the few patients whose disease chromosomes had shown ancient recombinations (6,11), the IOSCA locus could be refined to a 300 kb region between Genescan5 and FLJ23209exon3, these two SNPs have been identified by us when sequencing the critical region (exact locations in the clone AL133215 available if requested). An aberrant allele length of one patient, previously thought to represent the telomeric recombination border of the critical interval, proved to be a D10S1265-microsatellite marker mutation of two AC repeats in the patient in question and the actual region of interest to lie just telomeric to D10S1265 (Fig. 1).
Positional candidate genes and their analyses
By database searches, the critical IOSCA region in a completely sequenced clone (GenBank accession no. AL133215) was found to contain six positional candidate transcripts: C10orf6, which we had already characterized and analyzed in the patients' samples (GenBank accession nos AF460991 and AF460992) (12); semaphorin4g (sema4g) (GenBank accession no. NM_017893), the mouse homolog of which is involved in axonal path finding in neurogenesis and is located in the mouse chromosome 19 in a region synthenic to human chromosome 10q24 (13); C10orf2 encoding Twinkle and Twinky, the mutations of which we have previously reported to underlie the pathogenesis of autosomal dominant progressive external ophthalmoplegia (adPEO) (GenBank accession nos NM_021830, AF292004 and AF292005) (14); mitochondrial ribosomal protein L43 (MRPL43) (GenBank accession nos NM_032112, NM_176792 and NM_176793) completely overlapping with the 5′-untranslated region (UTR) of C10orf2 in the opposite DNA strand; a putative tumor suppressor gene (GenBank accession no. NM_032429) and a hypothetical cDNA FLJ23209 (GenBank accession no. NM_024895) (Fig. 2). All these genes were analyzed at both DNA and RNA levels in the patients: complete cDNAs were reverse transcription (RT)–polymerase chain reaction (PCR)-amplified from lymphoblastoid (LB) mRNA samples and thoroughly sequenced, the expression levels of the genes were studied with patient and control total brain RNA samples using semi-quantitative RT–PCR and all the exons and exon–intron junctions of each transcript, as well as most of the introns of sema4g, MRPL43 and C10orf2, were sequenced from genomic PCR products, the sequenced region covering ∼44 kb in total. In addition, one predicted gene encoding a homolog of a rat tricarboxylate carrier-like protein SFXN3 (GenBank accession no. NM_030971), located just telomeric to the flanking SNP FLJ23209exon3, was analyzed similarly. In addition to those reported in both public and commercial databases, only about a dozen SNPs were found in all the sequences analyzed, none of them being potentially pathogenic, the exclusion having a solid base on the patients' and the control subjects' genotypes. Conventional Southern blot hybridizations with a few separate probes of the genes listed earlier to various genomic restriction digests of some patients carrying the founder mutation in homozygous form, the patient being a compound heterozygote, and their parents did not show any DNA rearrangements (data not shown).
C10orf2 mutations in IOSCA
In the coding sequence of C10orf2, we identified two pathogenically relevant alterations: all the patients carrying at least traces of the founder haplotype were homozygous for an adenine to guanine missense point mutation at nucleotide 1708 (in Genbank accession no. AF292004 sequence) changing a tyrosine to a cysteine in exon 3 (Y508C). The patient carrying one founder IOSCA chromosome and one with a novel mutation was a compound heterozygote having the 1708A→G mutation in the maternal founder allele and a silent cytosine to thymine substitution in the exon 2 at nucleotide 1472 of the cDNA (Genbank accession no. AF292004), the last nucleotide in the codon encoding A429, in the paternal allele (Fig. 3).
The major 1708A→G mutation was analyzed by either direct sequencing or solid-phase minisequencing method in all 15 IOSCA families and a total of 712 Finnish control samples, as well as in 95 non-Finnish controls. No healthy individuals with a homozygous G/G genotype could be found among the IOSCA families or control subjects. Among the 712 Finnish controls, eight were found to be carriers of the 1708A→G mutation, the rest being homozygous for the wild-type (wt) allele. The carrier frequency of this major mutation varied considerably between the different parts of the country, consistent with the clustering of the disease carrying families to two distinct counties (Fig. 4). No non-Finnish mutation carriers could be found. The silent minor 1472C→T mutation was analyzed similarly in 207 Finnish and 95 foreign controls, and no C/T heterozygotes, nor T/T homozygotes, were detected.
Quantification of allelic expression
An individual being heterozygous for a coding region SNP (cSNP) enables the quantification of the relative amounts of transcripts derived from different alleles. Thus, to study the effects of the silent transition in the exon 2 of the patient 4 being a 1708G/1472T compound heterozygote and to detect possible imbalances in the expression levels, we applied solid-phase minisequencing method for the quantification of the allelic expression. In this patient, 72% of the steady-state LB mRNAs harbored Twinkle/TwinkyY508C mutation. His father, carrying the silent mutation, showed similar asymmetrical expression levels, however not being quantifiable with equal accuracy. This means that the expression of the allele carrying the silent 1472C→T transition was decreased, being about 2.6 times lower than that of normal. Thus, the majority of the transcripts were of the Y508C-mutant type in the patient and of the wt in his father. The phenomenon of asymmetrical allelic expression was observed by analyzing either point mutation (Table 1; Fig. 5).
Immunofluorescence studies
To study the subcellular targeting and stability of the TwinkleY508C and TwinkyY508C proteins, we halted the protein synthesis in the transiently transfected cells with cycloheximidine (CHX)-containing media for increasing time intervals, followed by mitochondrial staining and antibody detection. TwinkleY508C showed punctate mitochondrial fluorescence even after 4 h CHX treatment, indicative of it being associated with mitochondrial nucleoids as previously reported for wt Twinkle (14) and not being increasingly degraded. TwinkyY508C showed diffuse mitochondrial localization similar to wt Twinky, and no abnormal degradation could be observed either (Fig. 6). The silent 1472C→T transition, not changing an amino acid but affecting the transcriptional level, neither disturbed the mitochondrial localization nor decreased the stability of either protein (data not shown).
Analyses of mitochondrial DNA
As the symptoms in IOSCA resemble those of mitochondrial disorders, the mitochondrial enzyme function and stability of mitochondrial DNA (mtDNA) were previously studied in patients between 2 and 29 years of age (3). Neither defects in mitochondrial enzyme activity nor deletions or depletion of mitochondrial deoxyribonucleic acid mtDNA could be observed at that time point. As dominant C10orf2 mutations are associated with accumulation of multiple deletions of mtDNA (14), we repeated these experiments, the oldest patients analyzed now reaching their mid-40s, but no indications of mitochondrial dysfunction or mtDNA instability were found (Fig. 7).
DISCUSSION
Many human neurological disorders are a result of progressive degeneration of specific neuronal subpopulations, leading to a severe spectrum of clinical symptoms and eventually to premature death. Among them, hereditary spinocerebellar ataxia syndromes constitute one heterogenic subgroup, which has been investigated during the past few decades to identify the clinical, genetic, molecular and cellular events associated with distinct neurological disease entities. IOSCA represents one of the most severe forms of inherited ataxias with symptoms resulting from the progressive degeneration of the cerebellum, brain stem and spinal cord and from sensory axonal neuropathy. The pattern of neuronal degeneration having been described in detail in IOSCA (3,15), the disease entity forms an interesting target for investigating not only the pathogenic process, but also the basic neuronal pathways involved. The eventual aim of this study has been to identify the constitutive molecular genetic defect in IOSCA, providing the basis for further studies on the mutated gene and the corresponding protein(s) in normal and diseased states. We present here that the gene defective in IOSCA is C10orf2 encoding Twinkle, a mitochondrial replicative helicase, and a rarer splice variant Twinky with so far unknown function.
Having mapped the IOSCA locus to 10q24 (9), we first restricted the area by microsatellite marker-based haplotypes. However, the microsatellite-based haplotypes proved to be very misleading, as a putative recombination of one patient, initially assessed to telomerically restrict the critical region, actually represented a marker mutation of two AC repeats in length. This could only be detected after ordered SNP markers were available for genotyping. Thus, the locus extended into the telomeric direction, providing many more positional candidate genes to be analyzed in a 300 kb interval. Of these, the gene encoding Twinkle and Twinky contained two pathogenically relevant point mutations in the IOSCA patients: a substitution of an adenine to guanine leading to a Y508C amino acid change in the corresponding proteins and a silent cytosine to thymine transition affecting allelic expression levels. We analyzed hundreds of Finnish and foreign control alleles for both these mutations, establishing population frequencies consistent with the prevalence of the disease. Considering the major Y508C mutation in Finland, both the carrier frequency and the regional distribution of the heterozygous carriers were in good agreement when compared with the observed versus expected incidence of IOSCA patients born as well as their area of origin. Typical of a disorder of the Finnish disease heritage, no Y508C mutation carriers, not to mention Y508C homozygous patients, could be found abroad.
The novel minor mutation could be detected in a heterozygous form only in the compound heterozygous patient and his father, indicating it being a very rare and most probably recent event. This minor IOSCA mutation, a silent 1472C→T coding region transition, has no effect on the predicted polypeptides, but reduces the steady-state allelic transcription level. Consequently, the majority of the C10orf2 transcripts in the patient's cells encode the TwinkleY508C and TwinkyY508C mutant polypeptides. In agreement with this, in the cells of his heterozygote father the majority of the transcripts were derived from the non-variant allele. Allele-specific expression has been found to be relatively common for human disease genes (16,17), and in principle, different splice variant sensitivities or mRNA decay rates could mimic inequality in expression levels. However, it seems highly unlikely that the minor IOSCA allele would represent just an allele being commonly silenced or having splice variant-specific effects. First, the 1472C→T mutant allele is absent among roughly 600 control chromosomes analyzed, rendering it most unlikely, that it would represent a normal, harmless variant. Furthermore, transcripts encoding either splice variant, both containing the cSNP derived from the 1472C→T mutant allele, showed similar asymmetries in mRNA level. The basic underlying mechanisms of this allele having a lower steady-state transcript level remain unknown: no consensus binding sites around nucleotide 1472 were identified. The clinical phenotype of the A1708G/C1472C compound heterozygous patient is only minimally cognitively milder than that observed in the patients homozygous for the major mutation. It thus seems that minor amounts of normal C10orf2 transcripts are not sufficient to rescue the IOSCA phenotype caused by the Y508C mutation, but a full amount of mRNAs expressed from at least one normal allele is required to preserve the development of a healthy individual.
The IOSCA mutations result in defects of Twinkle, a mitochondrial hexameric ring DNA helicase with high homology to the bacteriophage T7 gene 4–protein, and a shorter splice variant Twinky with no characterized function (14). In humans, Twinkle has been localized to mitochondrial nucleoids, which are clusters of proteins associated with mitochondrial DNA, whereas Twinky can be detected diffusely inside mitochondria (14). Twinkle possesses 5′ to 3′ helicase activity and is required for the minimal replication apparatus for mtDNA in vitro, together with the mitochondrial DNA polymerase gamma (POLG) and the single-strand binding protein (mtSSB) (18,19). Therefore, Twinkle seems to play a crucial role in the mtDNA maintenance, but its other functions, as well as those of Twinky, remain to be clarified. We cannot yet determine whether the clinical phenotype caused by IOSCA mutations represents an outcome of the dysfunction of Twinkle or Twinky. By minisequencing, we have found that ∼80% of mRNAs transcribed from C10orf2 code for Twinkle and the remaining 20% from the shorter polypeptide Twinky in all tissues analyzed (Kaisu Nikali, unpublished data). The amount of Twinky mRNA being significant, the encoded protein presenting as a monomer when overexpressed, and its submitochondrial localization differing from that of Twinkle all suggest a distinct functional role for Twinky.
We have previously shown that dominant mutations of Twinkle underlie adPEO characterized by accumulation of multiple mtDNA deletions in the patients' muscle (14,20). PEO is a heterogeneous disease with differing spectra of symptoms and modes of inheritance observed in separate families. In addition to Twinkle, mutations causing PEO phenotype have been observed in adenine nucleotide transferase 1 (ANT1) (21) and POLG (22). Twinkle PEO manifests most often as a myopathy, but may be accompanied by, for example, psychiatric symptoms. The Twinkle defects in adPEO lead to mtDNA instability, possibly via to an enhanced dNTP breakdown or a dysfunction of a multimer containing both wt and mutant polypeptides (14).
Interestingly, all the symptoms of IOSCA patients have been reported in various PEO families separately, the onset of the PEO disorders however being in adulthood. Initially, because of the symptoms reminiscent of those observed in mitochondrial disorders, IOSCA was thought to represent one of them, but no defects in mitochondrial enzyme activities or instability of mtDNA could be observed in the patients, most of them examined during early childhood. The identification of the IOSCA mutations in C10orf2, mutated also in adPEO, prompted us to ensure that the IOSCA mutations have no mild dominant PEO effects, which had not been observed in the family history investigated back to the 16th century (3,11). Re-examination of some IOSCA parents, carrying one Y508C-mutant C10orf2 allele, showed no disease-related symptoms (K. Setälä, personal communication). We also re-examined the muscle mtDNA in the oldest IOSCA patients, now in their mid-30s, but no deletions or depletion of mtDNA could still be identified. In contrast to PEO, the IOSCA pathogenesis thus cannot be explained by mtDNA defects, but indicates that Y508C-mutant helicase can perform well in mtDNA replication. Furthermore, respiratory chain function and oxygen consumption of the Y508C-mutant tissues are normal (3). However, we cannot exclude the defects of mtDNA or mitochondrial dysfunction specific to neuronal subpopulations in IOSCA brain tissue. The pure central nervous system (CNS) phenotype and lack of muscle mitochondrial involvement in IOSCA thus suggest that Twinkle and Twinky play still uncovered roles specific to the CNS. The major mechanisms by which the Y508C alteration affects Twinkle/Twinky function and causes IOSCA phenotype remain largely unknown. The Y508C mutation does not disturb the subcellular localization of either Twinkle or Twinky, nor does it seem to decrease the half-life of the proteins. Conventional northern- and western-hybridization analyses with patient and control RNA samples and wt and mutant proteins, respectively, did not show differences in the steady-state level of either (data not shown). Although Twinkle and Twinky are ubiquitously expressed in human cells, IOSCA shows strictly neuronal phenotype, the neurodegeneration being restricted to few neuronal subpopulations, namely, the Purkinje, basket and granular cells of the cerebellar cortex and the neurons in the posterior horns of the spinal cord (3). It could be that the major IOSCA mutation affects a still uncharacterized Twinkle or Twinky protein function, or their interactions with one of their largely unknown partners, in a cell-type-specific manner.
The roles of proteins involved in mtDNA maintenance or mitochondrial function in general, defects of which can cause spinocerebellar degeneration and ataxia syndromes, are only starting to unravel. Among hereditary ataxia syndromes, IOSCA most closely resembles FRDA, the most common recessive ataxia worldwide, but in Finland possessing prevalence lower than that of IOSCA. In addition to the similar pattern of inheritance, clinical phenotype and CNS pathology, loss of peripheral large myelinated fibers represents prominent features in both diseases. The protein defective in FRDA, frataxin, encodes an evolutionally conserved mitochondrial protein involved in various aspects of iron and energy metabolism and the FRDA cells response insufficiently to oxidative stress (23–31). Considering all this, IOSCA patients interestingly have subtle disturbances in the heme metabolism, which is highly dependent on cellular iron and oxygen balance (3,6). A recently characterized recessive adult-onset ataxia of spinocerebellar type with sensory neuropathy and epilepsy caused by POLG mutations (mitochondrial recessive ataxia syndrome, MIRAS) (Anu Suomalainen, unpublished data) resembles IOSCA in its clinical and morphological findings, as well as in its minimal amount of mtDNA mutations and lack of mitochondrial myopathy. Furthermore, it shows that defects in both the polymerase and the helicase of the essential mtDNA replication machinery affect the spinocerebellar tract. These three mitochondrial ataxia types—IOSCA, FRDA and MIRAS—share the similar pattern of inheritance, clinical phenotype and CNS pathology and the loss of peripheral large myelinated fibers. The proteins interacting with Twinkle, frataxin or POLG still remain largely unknown. It will be exciting to see whether these three proteins are involved in the same metabolic pathways, defect of which causes distinct diseases sharing the spectra of neurological symptoms, or whether oxidative stress resulting from impaired mitochondrial function and toxic for neuronal cells is the mechanism in common for clinically similar syndromes.
The only non-neuronal manifestation in IOSCA is female primary hypogonadism of hypergonadotropic type, indicating a gonadal failure. Recently POLG mutations causing PEO phenotype and Parkinsonism were also linked to premature menopause (32). In these female PEO patients, high gonadotropin and low oestrogen levels were observed, implicating an ovarian dysfunction, just as in IOSCA. Previously, no Twinkle or Twinky mutations have been associated with hormone imbalance. However, these studies pose new questions not only about the functions of Twinkle and Twinky in the regulation of female hormone levels, but also the involvement of other mitochondrial proteins on the pathway in question. In addition, the recent findings of Hudson et al. (33) on a novel C10orf2 mutation leading to germ cell mosaicism, observed also in adPEO caused by ANT1 mutations (34), open new questions of the cellular roles of Twinkle and Twinky and the behavior of mitochondrial proteins in general.
To summarize, we describe here the first recessive disease phenotype caused by mutations in mitochondrial proteins, Twinkle and Twinky. Our findings join IOSCA, a devastating disorder with a specific pattern of neural degeneration, to the recently recognized group of spinocerebellar ataxias due to defects in mitochondrial enzyme functions. Twinkle has been previously shown to be essential for mtDNA maintenance as a replicative helicase, but IOSCA patients do not show signs of mtDNA instability. Our study thus opens new vistas of still uncharacterized roles of Twinkle and Twinky, probably most important or necessary only for the development and maintenance of the dying cerebellar and spinal neurons and sensory myelinated axons. Revealing the DNA-level background of IOSCA, this study forms the basis for further investigations of the IOSCA and ataxia pathogenesis in general and provides an interesting starting point for further research aimed at resolving the exact cellular functions of Twinkle and Twinky, the metabolic pathways they are involved in and how different mutations in the same gene result in two distinct disease states. Moreover, the study provides intelligence for exact patient, carrier and pre-natal diagnosis and genetic counselling for affected families.
MATERIALS AND METHODS
Study subjects and DNA and RNA sample preparation
All Finnish IOSCA patients diagnosed so far and their family members were included in the study. The family material has been described previously (1,6). Total DNA of the study subjects was extracted from leukocytes of peripheral venous blood samples, according to standard procedures (35). Total brain RNAs from a patient's and a control subject's frontal cortex autopsy samples were isolated using RNAeasy Mini Kit (Stratagene, LaJolla, CA, USA), according to the manufacturer's instruction, and the RNA samples were subsequently treated with RQ1 RNase-free DNase (Promega, Madison, WI, USA) to prevent DNA contamination. Messenger RNAs (mRNAs) from two patients', two parents' and a control subject's LB and fibroblastoid cell lines were extracted with FastTrack 2.0 mRNA Isolation Kit (Invitrogen, Leek, The Netherlands), following the manufacturer's recommendations.
PCR, RT–PCR and sequencing analyses
All the RT–PCR reactions were carried out with Titan One Step RT–PCR system (Roche Diagnostics, Espoo, Finland) as recommended by the manufacturer and with primers specific for each gene analyzed (primer sequences available on request). RT–PCR products were purified by shrimp alkaline phosphatase/exonuclease I treatment and sequenced with PCR and internal primers by ABI377 BigDye Terminator cycle sequencing protocol, according to the manufacturer's instruction (Perkin-Elmer). PCR reactions with genomic templates were performed with AmpliTaq Gold DNA polymerase (Applied Biosystems) in its buffer, and the primers were designed to amplify the exons as well as the exon–intron boundaries. Previously non-indicated exon–intron junctions were identified in advance by comparing the reported cDNA sequences with the genomic clone Genbank accession no. AL133215 using pairwise BLAST at the NCBI home page. The genomic amplification products were purified and sequenced as described earlier.
Genotyping of SNPs and haplotype construction
To refine the critical region of interest, four patients were chosen for SNP genotyping and haplotype construction: one patient carrying the ancient founder haplotype in its entirety in homozygous state, two patients showing ancient recombinations in either one of their founder disease chromosomes and one being heterozygous for the founder haplotype and suspected to carry a rare novel mutation in his paternal disease allele. SNPs around the microsatellite marker D10S1265 were searched for from both public and commercial databases and those reported to be polymorphic in at least two independent ones were selected for genotyping. PCR primers flanking the SNPs were used to amplify fragments ∼200–400 bp, and the amplification products were subsequently sequenced with PCR primers to monitor the genotypes of the patients. PCR and sequencing reactions were carried out as described earlier for genomic samples. Haplotypes were constructed manually on the basis of the genomic sequences available and assuming a minimum number of recombinations.
Analysis of positional candidate genes
Known genes and expressed sequence tag (EST) clusters located between Genscan5 and rs807014 were identified by database searches, potential exons on the interval were predicted by general exon prediction programs and full-length cDNAs were first constructed in silico by aligning all the sequences representing each separate transcript using both pairwise BLAST and Sequencher software (Gene Codes Corporation). All the genes were then RT–PCR-amplified and subsequently sequenced from both cDNA and genomic samples, respectively, of the patients and controls (primer sequences available on request). To exclude mutations affecting expression levels, semi-quantitative RT–PCR experiments were performed with patient and control total brain RNA samples as templates and using Titan One Step RT–PCR kit as described earlier.
Analyses of Twinkle/Twinky IOSCA mutations in the patient and control samples
After identifying the IOSCA mutations in the cDNA and genomic C10orf2 sequences of a few patients by sequencing RT–PCR products, mutation detection in all the Finnish IOSCA patients was carried out by PCR-amplifying the patients' and a few control subjects' DNA samples and subsequent direct sequencing of both DNA strands. The genotypes of the IOSCA families and those of the control subjects were then determined by direct sequencing and solid-phase minisequencing, respectively. In addition to the IOSCA parents and siblings, four collections of anonymous control DNAs were screened: 100 rural samples collected from Southern Botnia representing the early settlement region of Finland, another 100 samples from North Karelia representing the late settlement region (kind gifts from Dr Markus Perola), 500 samples collected from three different subpopulations of Finland and 100 non-Finnish European samples. The major mutation was analyzed in all control samples described earlier and the minor one in the early/late settlement samples as well as in the European controls.
Quantification of allelic expression
LB mRNAs from IOSCA patients numbered 1 and 4, as well as parents being heterozygous for either the major 1708A→G mutation or the minor 1472C→T mutation, and a control subject were used as templates when RT–PCR-amplifying Twinkle/Twinky cDNAs for minisequencing. RT–PCR reactions were carried out with Titan One Step RT–PCR system as recommended by the manufacturer, and the PCR-primer sequences used were TwEx5-R2 (Biotin-5′-GGAGAAGGTGAGGGAGTTCTTGTT-3′)+TwEx1F (5′-CTTTCCAGACCTCAATCGTATCTT-3′). Primer sequences for detecting the mutations in the exons 2 and 3 were 5′-GACGACATTCATCAGTGAGTATGC-3′ and 5′-CAATGCAACATGCAGTCTACGTCT-3′, respectively. The solid-phase minisequencing reactions were carried out essentially as described previously (36). The amount of mRNAs derived from different alleles was first estimated by calculating the ratios R=c.p.m. detecting the 1708A→G mutation/c.p.m. detecting wt nucleotide 1708A and R=c.p.m. detecting the wt nucleotide 1472C/c.p.m. detecting the mutant nucleotide 1472T. To more exactly determine the actual relative amounts of mRNAs transcribed from C10orf2, standard curves were constructed as previously described (36,37). In essence, the standard curves were prepared for both Twinkle and Twinky transcripts using a mixture of RNAs from an IOSCA patient (G/G homozygote) and a control subject (A/A homozygote). Genomic samples from two IOSCA parents being heterozygous (G/A) for the mutations served as control templates having both alleles equally represented. RT–PCR and minisequencing were carried out together with actual samples as described. When the acquired Twinkle/Twinky mRNA values were calibrated, a 12-point standard curve (y=0.014x−0.6739) was obtained (Table 1; Fig. 5). On the basis of this curve, the compound heterozygous patient's relative levels of expressed alleles of Twinkle/Twinky transcripts were determined more exactly.
Site-specific mutagenesis
1708A→G and 1472C→T mutation constructs were prepared with Quikchange Site-Directed Mutagenesis Kit (Stratagene), according to the manufacturer's instruction. FLAG-tagged wt-construct previously cloned in pCMV4 vector (Stratagene) (14) was used as a template, and the mutagenic primer sequences were as follows: 5′-GCAGTCTACGTCTGTGACATTTGTCATGTG-3′ and 5′-CACATGACAAATGTCACAGACGTAGACTGC-3′ for the TwinkleY508C and TwinkyY508C constructs and 5′-CATCAGTGAGTATGCTCTGGATTTGTGTTCCC-3′ and 5′-GGGAACACAAATCCAGAGCATACTCACTGATG-3′ for the 1472C→T transition in both splice variants. All the constructs were verified by direct sequencing.
Immunofluorescence
To study the subcellular localization and stability of the TwinkleY508C and TwinkyY508C proteins, baby hamster kidney-21 cells (CCL-10; ATCC, Manassas, USA) were cultured in Glasgow Modified Eagle Medium supplemented with 5% fetal calf serum, 10% tryptose-phosphate broth, 20 mm HEPES, glutamine and antibiotics. For transfections, the cells were seeded on six-well plates at a density of 3×105 cells per well and incubated overnight. Transient transfections were performed with the FuGENE6 transfection reagent (Roche Diagnostics) following the instructions of the manufacturer and using C-terminally in-frame FLAG-tagged pCMV4-plasmid constructs. Twenty-four hours post-transfection, the cells were plated on cover slips and the experiments were performed 48 h post-transfection. To halt the protein synthesis, the cells were incubated in the media containing 50 µg/ml CHX (Sigma, St Louis, MO, USA) for 0–4 h. Thereafter, the mitochondria were stained with MitoTracker Red CMXRos (Molecular Probes, Eugene, OR, USA) by incubating the cells in the media with the final MitoTracker Red concentration of 200 nm for 30 min. The fixation of the cells was carried out with 4% paraformaldehyde (PFA) for 20 min. PFA-fixed cells were then permeated with −20°C methanol. The cells were primarily labeled with mouse monoclonal anti-FLAG-IgG (Sigma), and FITC-conjugated secondary antibody was used for the final detection. After washes with phosphate-buffered saline and water, the cells were mounted in Gel/Mount (Biomeda, Foster City, CA, USA) and visualized using Leica DMR confocal immunofluorescence microscopy with TCS NT software.
Analyses of mitochondrial DNA
To study the integrity of mtDNA in the IOSCA patients' muscle, total DNA was extracted from muscle biopsy samples of four IOSCA patients of >30 years of age and controls, who were eventually diagnosed with other than muscle diseases, according to routine protocols. The whole mtDNA was PCR-amplified with Expand Long Template kit (Roche Diagnostics), using primers described by Tengan and Moraes (38). The amplification products were separated on ethidium bromide-containing agarose gels and inspected visually under ultraviolet light for aberrant products. Alternatively, 5 µg of total DNA was digested with restriction enzymes PvuII or BamHI (New England Biolabs) in their buffers and analyzed by Southern blot as described by Suomalainen et al. (20).
ELECTRONIC DATABASE INFORMATION
BLAST, http://www.ncbi.nlm.nih.gov/BLAST/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Prediction program, SNP database, genome map and transcript information links at http://www.ensembl.org/,http://www.ncbi.nlm.nih.gov/,http://www.hgsc.bcm.tmc.edu/,http://genome.ucsc.edu/, and http://www.celera.com/
Twinkle and Twinky GenBank accession nos AF292004 and AF292005, respectively.
ACKNOWLEDGEMENTS
The authors thank Ritva Timonen and Kati Ahlqvist for their skilful technical assistance, Drs Juha Isosomppi, Nabil Enattah and Jani Saarela for methodological help and fruitful discussions, Dr Kirsi Setälä for sharing her expertise as a neuro-ophthalmologist and Dr Leena Autio for Last Hope. The study was financially supported by the Emil Aaltonen Foundation (to K.N.), the Sigrid Juselius Foundation and the Biocentrum Helsinki (to L.P and A.S.) and the Centers of Excellence of Disease Genetics and FinMIT of the Academy of Finland (L.P. and A.S.).
Conflict of Interest statement. None of the authors have declared any conflict of interest.
Present address: MRC Developmental Neurobiology Centre, New Hunt's House, Guy's Hospital Campus, King's College, London SE1 1UL, UK.

Figure 1. The genotypes and haplotypes of the four IOSCA patients analyzed for restricting the critical region. The SNP and microsatellite markers used in genotyping are shown on the left, the total physical distance between the markers D10S603 and D10S1268 being 3.5 Mb (based on NCBI sequence contig NT_030059). The founder IOSCA haplotype is shown second on the left gray shaded and traces of it are indicated similarly in the patient haplotypes. Patient 1 is homozygous for the ancient founder haplotype in its entirety; patient 2 shows old telomeric recombinations in both of her disease chromosomes and a mutated D10S1265 marker allele; patient 3 shows a centromeric recombination on one of his founder disease chromosome and a telomeric one in the other and patient 4 is a compound heterozygote for the founder disease chromosome and an other carrying a novel mutation, this haplotype being totally different from the IOSCA founder haplotype. On the basis of these haplotypes, the IOSCA locus could be restricted to a 300 kb interval between markers Genscan5 and FLJ23209exon3. The subsequently identified IOSCA founder and novel mutations are shown boxed and circled, respectively, in each case (nd, genotype not determined).

Figure 2. A transcript map of the critical IOSCA interval. Simplified from the UCSC Human Genome Browser (May 2004 Assembly) chromosome 10 positions 102 500K–102 800K and the corresponding region in Ensembl Human Contig View. Not to exact scale.
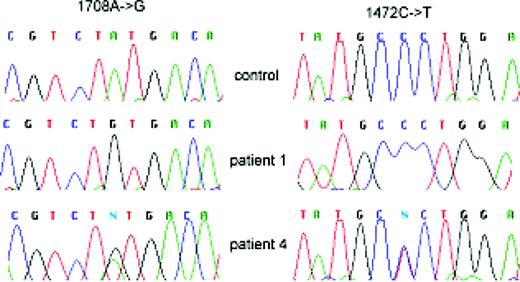
Figure 3. Sequence traces showing from top to bottom the genotypes of (i) a control subject homozygous for wt nucleotides 1708A and 1472C; (ii) patient 1 homozygous for the founder 1708A→G substitution mutation in the exon 3 and wt nucleotide 1472C and (iii) patient 4 heterozygous for the founder 1708A→G mutation and carrying a novel 1472C→T transition mutation in the exon 2 in his paternal chromosome. Column I, IOSCA major, i.e. 1708A→G mutation and column II, IOSCA minor, i.e. 1472C→T mutation.
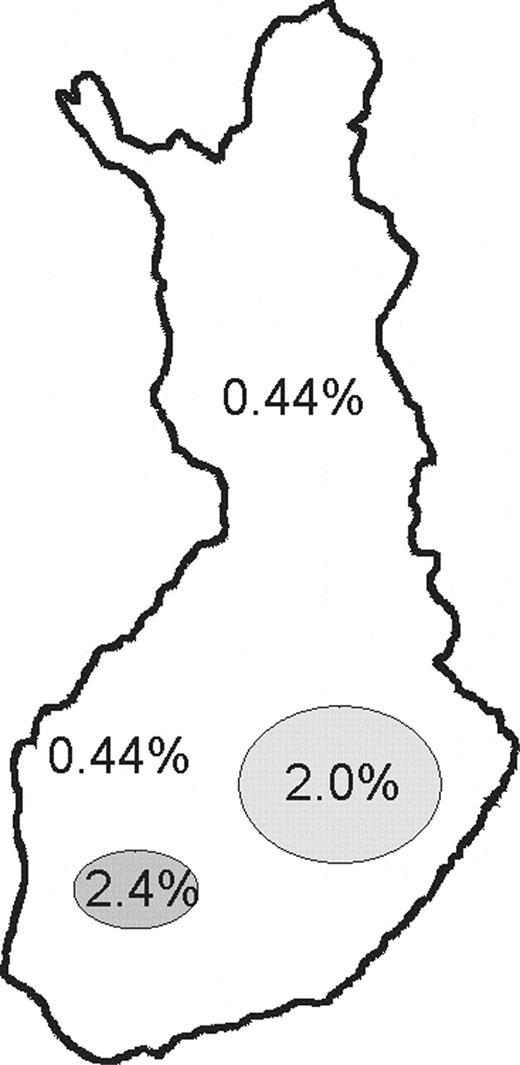
Figure 4. Carrier frequencies of the major IOSCA mutation in different parts of Finland. The regional clustering of heterozygous carriers can be logically observed in the areas where most of the IOSCA patients originate. In eastern Finland, where most of the IOSCA ancestors originate, the carrier frequency was 2%; in a regional subisolate in the county of Pirkanmaa, it was as high as 2.4%, most probably because of the segregation of only one disease chromosome in this isolate and in the rest of the country 0.44%.
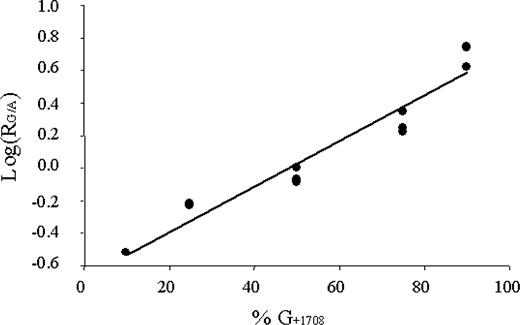
Figure 5. Standard curve constructed for exact determination of the amount of expressed alleles after solid-phase minisequencing. The amount of the major IOSCA allele is plotted as a function of G/A ratio on a log scale. This quantitative minisequencing analysis revealed that in the compound heterozygous patient most of the C10orf2 transcripts were derived from the Y508C mutation carrying allele, meaning that the IOSCA minor 1472C→T mutation substantially reduces allelic expression levels.
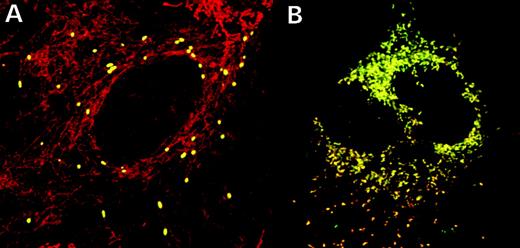
Figure 6. Expression studies of the mutant proteins in the case of both splice variants, shown as an overlay. The subcellular localization of the mutant proteins was studied by mitochondrial and immune co-staining and the half-life by exposure of the cells with CHX for varying time intervals, the longest one being 4 h. Even after 4 h incubation in CHX-containing media, the staining of cells transfected with either Twinkle (A) or Twinky (B) Y508C construct did not differ from what we have previously reported for wt-construct transfected cells: Twinkle being punctately localized in mitochondrial nucleoids and Twinky showing diffuse mitochondrial staining (14).
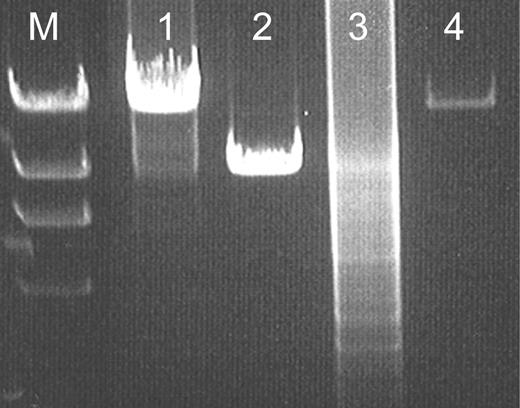
Figure 7. Long-range PCR analysis of one of the oldest IOSCA patient's mtDNA. Results from the analysis are shown in the following template order. Line M, DNA molecular weight marker lambda digested with HindIII, the longest digestion product being 16.6 kb; line 1, IOSCA patient; line 2, sporadic-adPEO patient having a single mtDNA deletion; line 3, familiar adPEO patient suffering from multiple mtDNA deletions and line 4, healthy control subject. In the IOSCA patient and control subject, only the full-length mtDNA of roughly 16.6 kb is amplified, a single deletion of mtDNA can be observed in a patient suffering from sporadic PEO, and the sample derived from an adPEO patient is smeared because of multiple deletions of mtDNA.
Relative amounts of the different C10orf2 alleles in control, homozygous IOSCA major and compound heterozygous IOSCA minor patient RNA samples, as well as heterozygous parents carrying either mutation
| Splice variant and mutation analyzed | Control | Patient 4 compound heterozygous for 1708A→G and 1472C→T mutations | Patient 4 compound heterozygous for 1708A→G quantitated allele ratio (%) | Parent heterozygous for 1472C→T transition | Patient 1 homozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation quantitatedallele ratio (%) |
|---|---|---|---|---|---|---|---|
| Twinkle G/A1708 | 0.05 | 2.32 | 74/26 | 0.04 | 47.32 | 1.1 | 51/49 |
| Twinkle C/T1472 | 56.62 | 2.6 | 2.35 | 14.22 | 13.51 | ||
| Twinky G/A1708 | 0.03 | 2.02 | 70/30 | 0.03 | 74.45 | 1.16 | 53/47 |
| Twinky C/T1472 | 33.91 | 2.25 | nd | 17.04 | 11.49 |
| Splice variant and mutation analyzed | Control | Patient 4 compound heterozygous for 1708A→G and 1472C→T mutations | Patient 4 compound heterozygous for 1708A→G quantitated allele ratio (%) | Parent heterozygous for 1472C→T transition | Patient 1 homozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation quantitatedallele ratio (%) |
|---|---|---|---|---|---|---|---|
| Twinkle G/A1708 | 0.05 | 2.32 | 74/26 | 0.04 | 47.32 | 1.1 | 51/49 |
| Twinkle C/T1472 | 56.62 | 2.6 | 2.35 | 14.22 | 13.51 | ||
| Twinky G/A1708 | 0.03 | 2.02 | 70/30 | 0.03 | 74.45 | 1.16 | 53/47 |
| Twinky C/T1472 | 33.91 | 2.25 | nd | 17.04 | 11.49 |
Genomic G/A1708=1.062 and genomic C/T1472=1.050. G/A (major) and C/T (minor) ratios in both splice variants are shown. The ratios implicate a strong decrease the steady-state expression levels of the allele containing the minor 1472C→T mutation, leading to the relative higher abundance of the mRNA encoding the mutant Y508C proteins in the compound heterozygous patient (numbered 4) and of the mRNA encoding the wt allele in his father. Genomic controls analyzed equal ratios close to 1.
Relative amounts of the different C10orf2 alleles in control, homozygous IOSCA major and compound heterozygous IOSCA minor patient RNA samples, as well as heterozygous parents carrying either mutation
| Splice variant and mutation analyzed | Control | Patient 4 compound heterozygous for 1708A→G and 1472C→T mutations | Patient 4 compound heterozygous for 1708A→G quantitated allele ratio (%) | Parent heterozygous for 1472C→T transition | Patient 1 homozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation quantitatedallele ratio (%) |
|---|---|---|---|---|---|---|---|
| Twinkle G/A1708 | 0.05 | 2.32 | 74/26 | 0.04 | 47.32 | 1.1 | 51/49 |
| Twinkle C/T1472 | 56.62 | 2.6 | 2.35 | 14.22 | 13.51 | ||
| Twinky G/A1708 | 0.03 | 2.02 | 70/30 | 0.03 | 74.45 | 1.16 | 53/47 |
| Twinky C/T1472 | 33.91 | 2.25 | nd | 17.04 | 11.49 |
| Splice variant and mutation analyzed | Control | Patient 4 compound heterozygous for 1708A→G and 1472C→T mutations | Patient 4 compound heterozygous for 1708A→G quantitated allele ratio (%) | Parent heterozygous for 1472C→T transition | Patient 1 homozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation | Parent heterozygous for 1708A→G mutation quantitatedallele ratio (%) |
|---|---|---|---|---|---|---|---|
| Twinkle G/A1708 | 0.05 | 2.32 | 74/26 | 0.04 | 47.32 | 1.1 | 51/49 |
| Twinkle C/T1472 | 56.62 | 2.6 | 2.35 | 14.22 | 13.51 | ||
| Twinky G/A1708 | 0.03 | 2.02 | 70/30 | 0.03 | 74.45 | 1.16 | 53/47 |
| Twinky C/T1472 | 33.91 | 2.25 | nd | 17.04 | 11.49 |
Genomic G/A1708=1.062 and genomic C/T1472=1.050. G/A (major) and C/T (minor) ratios in both splice variants are shown. The ratios implicate a strong decrease the steady-state expression levels of the allele containing the minor 1472C→T mutation, leading to the relative higher abundance of the mRNA encoding the mutant Y508C proteins in the compound heterozygous patient (numbered 4) and of the mRNA encoding the wt allele in his father. Genomic controls analyzed equal ratios close to 1.
References
Koskinen, T., Santavuori, P., Sainio, K., Lappi, M., Kallio, A.-K. and Pihko, H. (
Koskinen, T., Sainio, K., Rapola, J., Pihko, H. and Paetau, A. (
Lönnqvist, T. (
Norio, R., Nevanlinna, H.R. and Perheentupa, J. (
Peltonen, L., Jalanko, A. and Varilo, T. (
Nikali, K. (
Harding, A.E. (
Puccio, H. and Koenig, M. (
Nikali, K., Suomalainen, A., Terwilliger, J., Koskinen, T., Weissenbach, J. and Peltonen, L. (
Varilo, T., Nikali, K., Suomalainen, A., Lönnqvist, T. and Peltonen, L. (
Nikali, K., Isosomppi, J., Lönnqvist, T., Mao, J.I., Suomalainen, A. and Peltonen, L. (
Nikali, K., Saharinen, J. and Peltonen, L. (
Li, H., Wu, D.K. and Sullivan, S.L. (
Spelbrink, J.N., Li, F.Y., Tiranti, V., Nikali, K., Yuan, Q.P., Tariq, M., Wanrooij, S., Garrido, N., Comi, G., Morandi, L. et al. (
Lönnqvist, T., Paetau, A., Nikali, K., von Boguslawski, K. and Pihko, H. (
Yan, H., Yuan, W., Velculescu, V.E., Vogelstein, B. and Kinzler, K.W. (
Pastinen, T., Sladek, R., Gurd, S., Sammak, A., Ge, B., Lepage, P., Lavergne, K., Villeneuve, A., Gaudin, T., Brandstrom, H., et al. (
Korhonen, J.A., Gaspari, M. and Falkenberg, M. (
Korhonen, J.A., Pham, X.H., Pellegrini, M. and Falkenberg, M. (
Suomalainen, A., Majander, A., Wallin, M., Setala, K., Kontula, K., Leinonen, H., Salmi, T., Paetau, A., Haltia, M., Valanne, L. et al. (
Kaukonen, J., Juselius, J.K., Tiranti, V., Kyttala, A., Zeviani, M., Comi, G.P., Keranen, S., Peltonen, L and Suomalainen, A. (
Van Goethem, G., Luoma, P., Rantamaki, M., Al Memar, A., Kaakkola, S., Hackman, P., Krahe, R., Lofgren, A., Martin, J.J., De Jonghe, P. et al. (
Babcock, M., de Silva, D., Oaks, R., Davis-Kaplan, S., Jiralerspong, S., Montermini, L., Pandolfo, M. and Kaplan, J. (
Rötig, A., de Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., Munnich, A. and Rustin, P. (
Lodi, R., Cooper, J.M., Bradley, J.L., Manners, D., Styles, P., Taylor, D.J. and Shapira, A.H. (
Radisky, D.C., Babcock, M.C. and Kaplan, J. (
Wong, A., Yang, J., Cavadini, P., Gellera, C., Lonnerdal, B., Taroni, F. and Cortopassi, G. (
Bradley, J.L., Blake, J.C., Chamberlain, S., Thomas, P.K., Cooper, J.M. and Shapira, A.H. (
Wilson, R.B., Lynch, D.R., Farmer, J.M., Brooks, D.G. and Fischbeck, H.K. (
Foury, F. and Talibi, D. (
Scarano, V., de Cristofaro, T., de Michele, G., Salvatore, E., de Biase, I., Monticelli, A., Filla, A. and Cocozza, S. (
Luoma, P., Melber, A., Rinne, J.O., Kaukonen, J.A., Nupponen, N.N., Chalmers, R.M., Oldfors, A., Rautakorpi, I., Peltonen, L., Majamaa, K., Somer, H. and Suomalainen, A. (
Hudson, G., Deschauer, M., Busse, K., Zierz, S. and Chinnery, P.F. (
Deschauer, M., Hudson, G., Muller, T., Taylor, R.W., Chinnery, P.F and Zierz, S. (
Vandenplas, S., Wiid, I., Grobler-Rabie, A., Brebner, K., Ricketts, M., Wallis, G., Bester, A., Boyd, C. and Mathew, C. (
Suomalainen, A. and Syvänen, A.C. (
Kuokkanen, M., Enattah, N.S., Oksanen, A., Savilahti, E., Orpana, A. and Jarvela, I. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}