





Abstract
Background: Primary angiitis of the central nervous system (PACNS), a serious disease, has not featured prominently in the spectrum of multi-organ disease seen in vasculitis clinics.
Aim: To evaluate the presentation, natural history and features of PACNS variants and compare to those of systemic vasculitides.
Design: Retrospective analysis.
Methods: Patients (n = 105) presented during 1988–2003 to a tertiary regional vasculitis clinic receiving unselected disease types. Data were collected from a clinical database, patient and laboratory records.
Results: The frequency of PACNS presentation rose over the study period, compared with most of the other vasculitides. When PACNS was divided into small- and middle-sized vessel disease (SVD/MVD), their clinical courses differed substantially. SVD PACNS was responsive to immunosuppressive drugs, but relapsed during prolonged periods in all patients on maintenance immunosuppressives, or after withdrawal of treatment, causing recurrent, severe and irreversible CNS injury. MVD PACNS had isolated episodes at presentation, with a paucity of relapses during prolonged follow-up.
Discussion: Similarities between SVD PACNS and microscopic polyarteritis suggest the former may represent a limited form of the latter. MVD PACNS has a distinctly more benign relapse pattern than its multisystem counterpart polyarteritis nodosa. Acute-phase serology was useful in designating inflammatory processes at presentation of patients presenting with encephalopathy caused by SVD only, but were unhelpful in defining relapses in this form of PACNS, the definition of which in all cases rested on clinical assessment and MR scanning. Direct cerebral angiography was not diagnostic in any case of SVD PACNS; positive brain biopsy is diagnostically unequivocal, but the total clinical syndrome with imaging may establish a diagnosis with highest probability. In MVD PACNS, angiography with MR scan proved diagnostic. We suggest an algorithm for a rational, minimally invasive approach to investigation. In PACNS, SVD and MVD are important variants, and decisions about therapy should incorporate these distinctions.
Introduction
Primary angiitis of the central nervous system (PACNS) is a serious disease, potentially causing 3–5% of cerebrovascular accidents in patients aged <50 years,1 but the syndrome has not featured prominently in the spectrum of unselected, multi-organ disease seen in vasculitis clinics.2 Between 1959 and 1986, only 46 cases of PACNS had been described in the literature, most of these diagnosed at post-mortem.3 In 1991, Crane and Kerr illustrated 11 cases from the preceding decade,4 and more recently a six-patient series had magnetic resonance (MR) and angiographic findings delineated.5 Clinical experience suggests more patients suffering PACNS are now being recognized, but there are no data to evaluate whether this represents a true increase in incidence, or if it reflects insight and acumen in previously undiagnosed cases.
PACNS was not commonly recognized when the American College of Rheumatology (ACR)6 and Chapel Hill Consensus Conference (CHCC)7 criteria for the classification of primary systemic vasculitides were drawn up. Calabrese and Mallek suggested definitive criteria for the diagnosis of isolated angiitis of the central nervous system:3,8 (i) an acquired neurological deficit that remained unexplained after complete evaluation; (ii) a diagnostic cerebral angiogram that included diffuse areas of symmetric narrowing of vessels with areas of dilation and/or beaded vessel appearance, displacement of vessels or vessel occluded; (iii) no evidence of systemic vasculitis or any other condition that could mimic the angiogram findings.
This definition of PACNS excludes CNS disease where other vasculitic syndromes are partially or wholly identifiable and which encompass earlier descriptions that conferred heterogeneity in CNS disease by vessel size, or histologically, by presence or absence of granuloma.6,7 Diffuse cerebritis in small-vessel PACNS often manifests as subacute or acute encephalopathy, with persistent unexplained headache, altered higher mental function, confusion, and deteriorating conscious level, progressing to grand mal convulsions resistant to therapy, whereas middle-sized-vessel PACNS is more likely to present as a cerebrovascular accident in one or more defined or limited areas in patients without risk factors for arteriosclerotic disease. Central nervous system vasculitis is protean in its presentation, but enters the differential diagnosis of a patient when the pattern of disease is atypical, or when there are ischaemic infarcts, (occasionally haemorrhage8) temporally related, in separate and distinct vessel territories. Unifocal or absent MR findings do not exclude occult or more extensive CNS, multiple-vessel territory vasculitis9 which may be definable only on direct angiography of the CNS, in certain cases.
No therapeutic regimen for PACNS has been the subject of a controlled trial to assess comparative efficacies of drugs10 (corticosteroids, alone or in combination with cytotoxics) vs. risks of treatment, nor is this a likely prospect. A priori, the use of corticosteroids (with or without cytotoxic agents) to suppress disease activity in other organs, notably kidney and lung,11 and to sustain remission in the more common systemic vasculitides involving similar vessel types outside of the central nervous system (microscopic polyarteritis, polyarteritis nodosa, Wegener's granulomatosis) with similar vessel types within the CNS, seems a prudent initial approach to treatment. Recent descriptions of CNS vasculitis support the use of cytotoxics in addition to corticosteroids, but fall short of precisely how regimens should be managed in the longer term.12
Knowledge of the natural history of PACNS in long-term follow-up is critical in deciding optimal treatment regimens for patients. We therefore analysed data for patients presenting over a 15-year period to a tertiary regional vasculitis clinic, to identify patterns, organ injuries, natural history and features of PACNS compared to the systemic vasculitides.
Methods
The records of all patients attending the regional vasculitis clinic at Manchester Royal Infirmary between 1988 and 2003 were reviewed; 105 were included in the study. The final diagnoses identified were: polyarteritis nodosa (PAN); cutaneous vasculitis (variant, limited polyarteritis nodosa, or leucocytoclastic vasculitis) (CV); PACNS; Churg-Strauss syndrome (CSS); Wegener's granulomatosis (WG); microscopic polyarteritis (MPA); adult Henoch Schönlein purpura (HSP); and SLE vasculitis.
For inclusion in these groups, the patients had to meet the criteria set out by the ACR for PAN, CSS, WG, and SLE vasculitis; for HSP, an additional requirement of demonstration of typical glomerular mesangial deposition of immunoglobulin A was imposed. CHCC criteria were used for MPA and leukocytoclastic cutaneous vasculitis. For PACNS, Calabrese and Mallek's criteria were primary, using brain biopsy only when clinical and radiographic evidence was equivocal. The information abstracted is summarized in Table 1.
Case clinical and serological information abstracted
| Parameter | Data | |
|---|---|---|
| Year of diagnosis | 1988–2003 | |
| Route of referral | 10, 20, 30 care | |
| Organ involvement at presentation and during follow-up | Categories: cutaneous; ophthalmic; neurological; cardiac; pulmonary; gastrointestinal; renal; musculoskeletal | |
| Laboratory investigations | Acute-phase parameters: CRP, ESR | |
| Immunoglobulins: G, A, M, E, levels | ||
| Differential white cell counts; absolute eosinophils counts | ||
| Autoantibodies: anticardiolipin, lupus anticoagulant, c- ANCA, p-ANCA, ANF, dsDNA | ||
| Radiological imaging and diagnostic biopsies | Direct and/or MR enhanced angiography; CT and MR scanning of affected organs, including CNS when justified | |
| Treatments for PACNS | ||
| Induction therapy | Prednisolone 60 mg/day, tapering to 20 mg, with cyclophosphamide 1.5 mg/kg/day for 3 months | |
| Maintenance | Prednisolone tapering to nil, with azathioprine 1.5 mg/kg/day for 2 years | |
| Re-induction | As for induction | |
| Relapses, disease outcome clinical parameters, side-effects of therapy | Defined clinically primarily, supported by objective imaging and/or neurophysiology | |
| Parameter | Data | |
|---|---|---|
| Year of diagnosis | 1988–2003 | |
| Route of referral | 10, 20, 30 care | |
| Organ involvement at presentation and during follow-up | Categories: cutaneous; ophthalmic; neurological; cardiac; pulmonary; gastrointestinal; renal; musculoskeletal | |
| Laboratory investigations | Acute-phase parameters: CRP, ESR | |
| Immunoglobulins: G, A, M, E, levels | ||
| Differential white cell counts; absolute eosinophils counts | ||
| Autoantibodies: anticardiolipin, lupus anticoagulant, c- ANCA, p-ANCA, ANF, dsDNA | ||
| Radiological imaging and diagnostic biopsies | Direct and/or MR enhanced angiography; CT and MR scanning of affected organs, including CNS when justified | |
| Treatments for PACNS | ||
| Induction therapy | Prednisolone 60 mg/day, tapering to 20 mg, with cyclophosphamide 1.5 mg/kg/day for 3 months | |
| Maintenance | Prednisolone tapering to nil, with azathioprine 1.5 mg/kg/day for 2 years | |
| Re-induction | As for induction | |
| Relapses, disease outcome clinical parameters, side-effects of therapy | Defined clinically primarily, supported by objective imaging and/or neurophysiology | |
Case clinical and serological information abstracted
| Parameter | Data | |
|---|---|---|
| Year of diagnosis | 1988–2003 | |
| Route of referral | 10, 20, 30 care | |
| Organ involvement at presentation and during follow-up | Categories: cutaneous; ophthalmic; neurological; cardiac; pulmonary; gastrointestinal; renal; musculoskeletal | |
| Laboratory investigations | Acute-phase parameters: CRP, ESR | |
| Immunoglobulins: G, A, M, E, levels | ||
| Differential white cell counts; absolute eosinophils counts | ||
| Autoantibodies: anticardiolipin, lupus anticoagulant, c- ANCA, p-ANCA, ANF, dsDNA | ||
| Radiological imaging and diagnostic biopsies | Direct and/or MR enhanced angiography; CT and MR scanning of affected organs, including CNS when justified | |
| Treatments for PACNS | ||
| Induction therapy | Prednisolone 60 mg/day, tapering to 20 mg, with cyclophosphamide 1.5 mg/kg/day for 3 months | |
| Maintenance | Prednisolone tapering to nil, with azathioprine 1.5 mg/kg/day for 2 years | |
| Re-induction | As for induction | |
| Relapses, disease outcome clinical parameters, side-effects of therapy | Defined clinically primarily, supported by objective imaging and/or neurophysiology | |
| Parameter | Data | |
|---|---|---|
| Year of diagnosis | 1988–2003 | |
| Route of referral | 10, 20, 30 care | |
| Organ involvement at presentation and during follow-up | Categories: cutaneous; ophthalmic; neurological; cardiac; pulmonary; gastrointestinal; renal; musculoskeletal | |
| Laboratory investigations | Acute-phase parameters: CRP, ESR | |
| Immunoglobulins: G, A, M, E, levels | ||
| Differential white cell counts; absolute eosinophils counts | ||
| Autoantibodies: anticardiolipin, lupus anticoagulant, c- ANCA, p-ANCA, ANF, dsDNA | ||
| Radiological imaging and diagnostic biopsies | Direct and/or MR enhanced angiography; CT and MR scanning of affected organs, including CNS when justified | |
| Treatments for PACNS | ||
| Induction therapy | Prednisolone 60 mg/day, tapering to 20 mg, with cyclophosphamide 1.5 mg/kg/day for 3 months | |
| Maintenance | Prednisolone tapering to nil, with azathioprine 1.5 mg/kg/day for 2 years | |
| Re-induction | As for induction | |
| Relapses, disease outcome clinical parameters, side-effects of therapy | Defined clinically primarily, supported by objective imaging and/or neurophysiology | |
Data were entered into an Excel spreadsheet and analysed using the Stats Direct package. Analyses used the Poisson regression model to calculate the relative risks (e.g. the change in the rate of incidence) for each disorder with respect to self. χ2 for linear trend was used to evaluate the absolute rate of change for referral source over time in relation to the total study population. Fisher's exact test was used to analyse neurological involvement over time.
Results
We included 105 patients in the study. The primary diagnoses are shown in Table 2. Overall, the genders were balanced (M:F 1:0.98), but subgroups had apparent gender bias. The mean age was 46.2 years, the median 46.5 years.
Patient diagnoses and case mix
| Diagnosis | n | % of total | M:F |
|---|---|---|---|
| Polyarteritis nodosa | 17 | 16.2 | 8:9 |
| PACNS | 12 | 10.5 | 3:4 |
| Microscopic polyarteritis | 11 | 10.5 | 8:3 |
| Churg-Strauss syndrome | 11 | 10.5 | 8:3 |
| Adult Henoch Schönlein purpura | 14 | 13.3 | 9:5 |
| Wegener's granulomatosis | 20 | 19.0 | 3:2 |
| SLE vasculitis | 15 | 14.3 | 4:11 |
| Cutaneous vasculitis | 4 | 3.8 | 0:4 |
| Diagnosis | n | % of total | M:F |
|---|---|---|---|
| Polyarteritis nodosa | 17 | 16.2 | 8:9 |
| PACNS | 12 | 10.5 | 3:4 |
| Microscopic polyarteritis | 11 | 10.5 | 8:3 |
| Churg-Strauss syndrome | 11 | 10.5 | 8:3 |
| Adult Henoch Schönlein purpura | 14 | 13.3 | 9:5 |
| Wegener's granulomatosis | 20 | 19.0 | 3:2 |
| SLE vasculitis | 15 | 14.3 | 4:11 |
| Cutaneous vasculitis | 4 | 3.8 | 0:4 |
PACNS, primary angiitis of the central nervous system.
Patient diagnoses and case mix
| Diagnosis | n | % of total | M:F |
|---|---|---|---|
| Polyarteritis nodosa | 17 | 16.2 | 8:9 |
| PACNS | 12 | 10.5 | 3:4 |
| Microscopic polyarteritis | 11 | 10.5 | 8:3 |
| Churg-Strauss syndrome | 11 | 10.5 | 8:3 |
| Adult Henoch Schönlein purpura | 14 | 13.3 | 9:5 |
| Wegener's granulomatosis | 20 | 19.0 | 3:2 |
| SLE vasculitis | 15 | 14.3 | 4:11 |
| Cutaneous vasculitis | 4 | 3.8 | 0:4 |
| Diagnosis | n | % of total | M:F |
|---|---|---|---|
| Polyarteritis nodosa | 17 | 16.2 | 8:9 |
| PACNS | 12 | 10.5 | 3:4 |
| Microscopic polyarteritis | 11 | 10.5 | 8:3 |
| Churg-Strauss syndrome | 11 | 10.5 | 8:3 |
| Adult Henoch Schönlein purpura | 14 | 13.3 | 9:5 |
| Wegener's granulomatosis | 20 | 19.0 | 3:2 |
| SLE vasculitis | 15 | 14.3 | 4:11 |
| Cutaneous vasculitis | 4 | 3.8 | 0:4 |
PACNS, primary angiitis of the central nervous system.
PACNS vs. other vasculitides
Presentation frequencies were assessed by year of diagnosis. Overall, there was an increase in the relative rate of presentation of systemic vasculitis for each 5-year period from 1988 to 2003. Using Poisson regression, the presentation rate ratio (PRR) of the total number of patients across the three time groups was 1.336 (99% CI 0.975–1.829) (p = 0.017). (Figure 1), where a value of 1 indicates no change and a value of 1.5 indicates a 50% increase in the presentation rate.
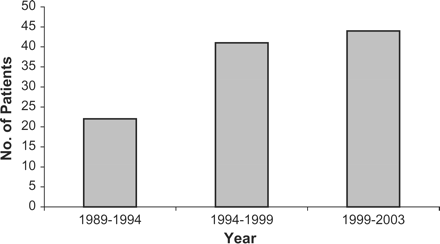
Patient total numbers diagnosed with systemic vasculitis in 5 year periods, 1988–2003.
PACNS only presented in the last 6 years of the study (1997–2003). Further analysis (Figure 2) showed a PRR for PACNS of 5.605 (99% CI 0.958–32.79) (p = 0.012), with a sex ratio of M:F 3:4.
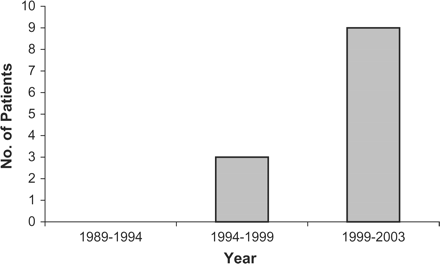
Presentations of primary angiitis of the central nervous system in 5-year periods 1998–2003.
In contrast, presentation frequency data for the other systemic vasculitides (not shown), revealed no significant change for WG (p = 0.2), HSP (p = 0.9), CV (p = 0.9), CSS (p = 0.4) and SLE secondary vasculitis (p = 0.5). MPA numbers fell, but not significantly so (p = 0.15). Only PAN showed a progressive increase (PRR 2.72, p = 0.004).
For all the systemic vasculitides taken together, 23% of patients had neurological involvement in primary and secondary CNS, and peripheral nervous system disease at the time of diagnosis (Figure 3). Patients with CSS, WG, MPA, PAN and SLE vasculitis, had varying frequencies of neurological disease, in addition to those with PACNS. These were peripheral, rather than central, nervous system involvement. Headaches were the commonest initial symptom before presentation of CNS disease (30%), accompanied by paraesthesia, with clinical evidence of hemiplegia, seizures, confusion, ataxia and signs of spasticity. Ten percent had peripheral neuropathy and 3% had mononeuritis multiplex.
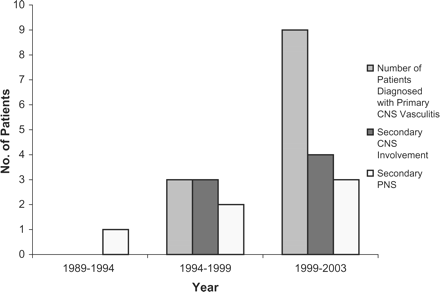
All patients with neurological disease: primary and secondary CNS, and peripheral nervous system disease.
The first case of diagnosed secondary peripheral nervous system involvement within the systemic vasculitides was in 1989. After 1994, there has been an increase in central nervous system involvement. From 1998, PACNS has been diagnosed in 12 patients, and the annual frequency (Figure 4) has been consistent over the past 5 years.
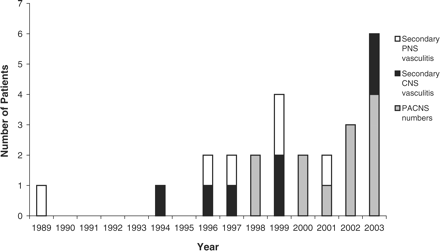
Annual presentations of neurological pathologies.
Laboratory findings
See Table 3. Weakly positive ANF (with negative dsDNA), c-ANCA, and p-ANCA autoantibodies were found in 14–29% of PACNS patients, but titres did not exceed 1:40 in any patient, and may have been a consequence of polyclonal activation associated with the acute phase response in patients. Acute-phase parameters at presentation showed two distinct patterns, according to whether the patient suffered PACNS of small-vessel type (5/6 raised), or middle vessel type (5/6 normal) (χ2 = 5.33; p < 0.02). These findings contrasted with acute-phase parameters during relapse (SVD cases only), when ESR and CRP remained normal in all instances.
Laboratory findings of significance in the systemic vasculitides and PACNS
| Diagnosis… | PACNS | PAN | CSS | HSP | MPA | WG | SLE | Cutaneous | Total |
|---|---|---|---|---|---|---|---|---|---|
| Laboratory findings | (%) | (%) | (%) | (%) | (%) | (%) | vasculitis (%) | vasculitis (%) | (%) |
| Raised IgE (>150 units) | 29 | 12 | 64 | – | – | – | 7 | – | 17 |
| Eosinophilia (>0.4 × 109/l) | – | – | 100 | – | – | 55 | – | – | 11 |
| Anti-cardiolipin (>16) | – | – | – | 7 | – | – | 7 | – | 2 |
| Lupus-anticoagulant | – | – | – | – | – | – | 93 | – | 14 |
| c-ANCA (>1/10) | 14 | 6 | 9 | – | – | 60 | – | – | 15 |
| p-ANCA (>1/10) | 29 | 12 | 9 | 7 | 55 | – | – | – | 12 |
| ANA (>1/40) | 14 | 18 | – | 7 | 9 | 5 | 13 | – | 9 |
| Diagnosis… | PACNS | PAN | CSS | HSP | MPA | WG | SLE | Cutaneous | Total |
|---|---|---|---|---|---|---|---|---|---|
| Laboratory findings | (%) | (%) | (%) | (%) | (%) | (%) | vasculitis (%) | vasculitis (%) | (%) |
| Raised IgE (>150 units) | 29 | 12 | 64 | – | – | – | 7 | – | 17 |
| Eosinophilia (>0.4 × 109/l) | – | – | 100 | – | – | 55 | – | – | 11 |
| Anti-cardiolipin (>16) | – | – | – | 7 | – | – | 7 | – | 2 |
| Lupus-anticoagulant | – | – | – | – | – | – | 93 | – | 14 |
| c-ANCA (>1/10) | 14 | 6 | 9 | – | – | 60 | – | – | 15 |
| p-ANCA (>1/10) | 29 | 12 | 9 | 7 | 55 | – | – | – | 12 |
| ANA (>1/40) | 14 | 18 | – | 7 | 9 | 5 | 13 | – | 9 |
Laboratory findings of significance in the systemic vasculitides and PACNS
| Diagnosis… | PACNS | PAN | CSS | HSP | MPA | WG | SLE | Cutaneous | Total |
|---|---|---|---|---|---|---|---|---|---|
| Laboratory findings | (%) | (%) | (%) | (%) | (%) | (%) | vasculitis (%) | vasculitis (%) | (%) |
| Raised IgE (>150 units) | 29 | 12 | 64 | – | – | – | 7 | – | 17 |
| Eosinophilia (>0.4 × 109/l) | – | – | 100 | – | – | 55 | – | – | 11 |
| Anti-cardiolipin (>16) | – | – | – | 7 | – | – | 7 | – | 2 |
| Lupus-anticoagulant | – | – | – | – | – | – | 93 | – | 14 |
| c-ANCA (>1/10) | 14 | 6 | 9 | – | – | 60 | – | – | 15 |
| p-ANCA (>1/10) | 29 | 12 | 9 | 7 | 55 | – | – | – | 12 |
| ANA (>1/40) | 14 | 18 | – | 7 | 9 | 5 | 13 | – | 9 |
| Diagnosis… | PACNS | PAN | CSS | HSP | MPA | WG | SLE | Cutaneous | Total |
|---|---|---|---|---|---|---|---|---|---|
| Laboratory findings | (%) | (%) | (%) | (%) | (%) | (%) | vasculitis (%) | vasculitis (%) | (%) |
| Raised IgE (>150 units) | 29 | 12 | 64 | – | – | – | 7 | – | 17 |
| Eosinophilia (>0.4 × 109/l) | – | – | 100 | – | – | 55 | – | – | 11 |
| Anti-cardiolipin (>16) | – | – | – | 7 | – | – | 7 | – | 2 |
| Lupus-anticoagulant | – | – | – | – | – | – | 93 | – | 14 |
| c-ANCA (>1/10) | 14 | 6 | 9 | – | – | 60 | – | – | 15 |
| p-ANCA (>1/10) | 29 | 12 | 9 | 7 | 55 | – | – | – | 12 |
| ANA (>1/40) | 14 | 18 | – | 7 | 9 | 5 | 13 | – | 9 |
Natural history of the PACNS forms
The time course of PACNS was analysed. Relapse was defined clinically as significant, sustained, new symptoms and signs during follow-up to 6 years from presentation. In all cases, evidence of further CNS infarcts was found on repeat imaging (Table 1).
In the 12 patients with PACNS, there were a total of 16 relapses: 12 single relapses, and four recurrent relapses. All of these relapses occurred in the six patients who had characteristics of diffuse small-vessel cerebral vasculitis. Relapses occurred in half of the patients whilst they were receiving low-dose maintenance immunosuppressives (prednisolone 10 mg/day or less, with azathioprine 1.5 mg/kg/day) and in others, 3–36 months after discontinuing immunosuppressive treatment. When PACNS was sub-classified into small- and large-vessel disease variants (SVD vs. MVD), the SVD variant was far more likely to relapse. No patient classified as MVD suffered a second active disease episode (Figure 5).
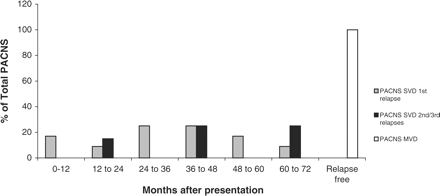
First and second clinical relapses of primary angiitis of the central nervous system during 6-year follow-up of small-vessel disease (SVD), and middle-size vessel disease (MVD).
PACNS radiographic characteristics
Radiological imaging was most useful in the diagnosis of PACNS, but in this study, direct angiography was helpful only in the MVD variant (Table 4), and CT scan clearly inferior to MR in both variants. Radiology is a widely accepted method of diagnosis in PACNS.1,13–17 Detailed presentation of radiographic features in this series is intended as the subject of an additional, complementary, paper.
Primary angiitis of the central nervous system (PACNS): patient characteristics: clinical and radiographic features, small (SVD) or middle-sized vessel disease (MVD), and natural disease history of clinical disease relapses
| Patient (age, sex) | PACNS type | Clinical presentation | Presentation acute phase parameters | CSF | CT | MR* | Angiography* | PACNS diagnostic criteria† |
|---|---|---|---|---|---|---|---|---|
| 1 (55M) | SVD | Confusion, headache, focal seizures, dysphasia | ESR 50, CRP 20 | Protein 0.7 g/l; cell count normal. Negative oligoclonal bands | Cerebral atrophy; dilation ventricles and sulci | Multifocal grey and white matter infarcts: right caudate nucleus, lateral ventricles: Relapses: new abnormalities | Peripheral small vessels hypoperfused | (iii) (iv) (v) |
| 2 (22F) | SVD | Headaches, grand mal seizures | Normal | Normal protein, cell count | Normal | Multiple white and grey matter infarcts* | Abnormal (venous phase) | (ii) (iii) (iv) (v) |
| 3 (32F) | SVD | Hemiparesis, encephalitic | ESR 32, CRP 50 | Normal; negative for oligoclonal bands at presentation and relapse | Normal | Multiple grey and white matter infarcts. Relapses: new MR abnor- mality | Normal | (iii) (iv) (v) |
| 4 (51F) | SVD | Migraines, grand mal seizures | ESR 38, CRP 55 | Protein 0.79 g/l; normal cell count | Acute lacunar infarct, left internal capsule | Multiple grey and white matter infarcts | Normal | (iii) (iv) (v) |
| 5 (53F) | SVD | Encephalitic, grand mal seizures | ESR 80, CRP 40 | Protein (0.74 g/l); WCC 20 × 106/l; 90% lymphocytes; RBC 4 × 106/l | Normal | Pulvinar sign* | (Not performed) | (i) (Brain biopsy) (iv) (v) |
| 6 (27M) | SVD | Lateral medullary syndrome | ESR 30, CRP 30 | Traumatic tap: normal WCC differential/RBC ratio | Normal | Multiple grey and white matter infarcts. Four relapses: new MR abnormalities | (iii) (iv) (v) | |
| 7 (16F) | MVD | Anton-Babinski syndrome | Normal | Normal cell count, protein | Normal at presen- tation; repeat: occipital infarct | Multiple high signals; cortical gyri, both occipitals; pre-, post-gyri, temporal, thalamus | Occluded central arteries: occipital poles bilaterally: gross luxury perfusion round occipital infarct | (ii) (v) |
| 8 (29F) | MVD | Aphasia, progressive hemiparesis | Normal | Normal | Normal | * | Characteristic vasculitic shoulder: progression on repeat images | (ii) (v) |
| 9 (28M) | MVD | Headache, ipsilateral cerebellar signs, hemiparesis, childhood polymyositis | Normal | Normal | Normal | Infarcts in cerebellum and contralateral internal capsule | Minor peripheral vessel abnormality | (iii) (v) |
| 10 (48M) | MVD | Occipital headache, cerebellar CVA | Normal | Not performed | Evolving left cerebellar infarct | * | Multiple vessels: characteristic vasculitis, extending beyond affected territories on MR | (ii) (iii) (v) |
| 11 (32M) | MVD | Quadriparesis, grand mal seizures | ESR 70, CRP 206 | Normal | Normal | Multiple vessel territory infarcts | Multiple vessels: characteristic vasculitis, | (ii) (iii) (v) |
| 12 (29F) | MVD | Aphasia, hemiplegia | Normal | Normal | Normal | Middle cerebral infarct | Internal carotid: vasculitis, progressing on repeat images | (ii) (iii) (v) |
| Patient (age, sex) | PACNS type | Clinical presentation | Presentation acute phase parameters | CSF | CT | MR* | Angiography* | PACNS diagnostic criteria† |
|---|---|---|---|---|---|---|---|---|
| 1 (55M) | SVD | Confusion, headache, focal seizures, dysphasia | ESR 50, CRP 20 | Protein 0.7 g/l; cell count normal. Negative oligoclonal bands | Cerebral atrophy; dilation ventricles and sulci | Multifocal grey and white matter infarcts: right caudate nucleus, lateral ventricles: Relapses: new abnormalities | Peripheral small vessels hypoperfused | (iii) (iv) (v) |
| 2 (22F) | SVD | Headaches, grand mal seizures | Normal | Normal protein, cell count | Normal | Multiple white and grey matter infarcts* | Abnormal (venous phase) | (ii) (iii) (iv) (v) |
| 3 (32F) | SVD | Hemiparesis, encephalitic | ESR 32, CRP 50 | Normal; negative for oligoclonal bands at presentation and relapse | Normal | Multiple grey and white matter infarcts. Relapses: new MR abnor- mality | Normal | (iii) (iv) (v) |
| 4 (51F) | SVD | Migraines, grand mal seizures | ESR 38, CRP 55 | Protein 0.79 g/l; normal cell count | Acute lacunar infarct, left internal capsule | Multiple grey and white matter infarcts | Normal | (iii) (iv) (v) |
| 5 (53F) | SVD | Encephalitic, grand mal seizures | ESR 80, CRP 40 | Protein (0.74 g/l); WCC 20 × 106/l; 90% lymphocytes; RBC 4 × 106/l | Normal | Pulvinar sign* | (Not performed) | (i) (Brain biopsy) (iv) (v) |
| 6 (27M) | SVD | Lateral medullary syndrome | ESR 30, CRP 30 | Traumatic tap: normal WCC differential/RBC ratio | Normal | Multiple grey and white matter infarcts. Four relapses: new MR abnormalities | (iii) (iv) (v) | |
| 7 (16F) | MVD | Anton-Babinski syndrome | Normal | Normal cell count, protein | Normal at presen- tation; repeat: occipital infarct | Multiple high signals; cortical gyri, both occipitals; pre-, post-gyri, temporal, thalamus | Occluded central arteries: occipital poles bilaterally: gross luxury perfusion round occipital infarct | (ii) (v) |
| 8 (29F) | MVD | Aphasia, progressive hemiparesis | Normal | Normal | Normal | * | Characteristic vasculitic shoulder: progression on repeat images | (ii) (v) |
| 9 (28M) | MVD | Headache, ipsilateral cerebellar signs, hemiparesis, childhood polymyositis | Normal | Normal | Normal | Infarcts in cerebellum and contralateral internal capsule | Minor peripheral vessel abnormality | (iii) (v) |
| 10 (48M) | MVD | Occipital headache, cerebellar CVA | Normal | Not performed | Evolving left cerebellar infarct | * | Multiple vessels: characteristic vasculitis, extending beyond affected territories on MR | (ii) (iii) (v) |
| 11 (32M) | MVD | Quadriparesis, grand mal seizures | ESR 70, CRP 206 | Normal | Normal | Multiple vessel territory infarcts | Multiple vessels: characteristic vasculitis, | (ii) (iii) (v) |
| 12 (29F) | MVD | Aphasia, hemiplegia | Normal | Normal | Normal | Middle cerebral infarct | Internal carotid: vasculitis, progressing on repeat images | (ii) (iii) (v) |
ESR, erythrocyte sedimention rate (mm/h); CRP, C-reactive protein (mg/l). *Findings displayed in Figures 7–10. †Diagnostic criteria for PACNS: (i) brain biopsy; (ii) characteristic angiographic findings; (iii) multiple CNS artery territory white and grey matter infarcts temporally related; (iv) relapsing course with new infarcts found on MR; (v) clinical response(s) to immunosuppressive regimen.
Primary angiitis of the central nervous system (PACNS): patient characteristics: clinical and radiographic features, small (SVD) or middle-sized vessel disease (MVD), and natural disease history of clinical disease relapses
| Patient (age, sex) | PACNS type | Clinical presentation | Presentation acute phase parameters | CSF | CT | MR* | Angiography* | PACNS diagnostic criteria† |
|---|---|---|---|---|---|---|---|---|
| 1 (55M) | SVD | Confusion, headache, focal seizures, dysphasia | ESR 50, CRP 20 | Protein 0.7 g/l; cell count normal. Negative oligoclonal bands | Cerebral atrophy; dilation ventricles and sulci | Multifocal grey and white matter infarcts: right caudate nucleus, lateral ventricles: Relapses: new abnormalities | Peripheral small vessels hypoperfused | (iii) (iv) (v) |
| 2 (22F) | SVD | Headaches, grand mal seizures | Normal | Normal protein, cell count | Normal | Multiple white and grey matter infarcts* | Abnormal (venous phase) | (ii) (iii) (iv) (v) |
| 3 (32F) | SVD | Hemiparesis, encephalitic | ESR 32, CRP 50 | Normal; negative for oligoclonal bands at presentation and relapse | Normal | Multiple grey and white matter infarcts. Relapses: new MR abnor- mality | Normal | (iii) (iv) (v) |
| 4 (51F) | SVD | Migraines, grand mal seizures | ESR 38, CRP 55 | Protein 0.79 g/l; normal cell count | Acute lacunar infarct, left internal capsule | Multiple grey and white matter infarcts | Normal | (iii) (iv) (v) |
| 5 (53F) | SVD | Encephalitic, grand mal seizures | ESR 80, CRP 40 | Protein (0.74 g/l); WCC 20 × 106/l; 90% lymphocytes; RBC 4 × 106/l | Normal | Pulvinar sign* | (Not performed) | (i) (Brain biopsy) (iv) (v) |
| 6 (27M) | SVD | Lateral medullary syndrome | ESR 30, CRP 30 | Traumatic tap: normal WCC differential/RBC ratio | Normal | Multiple grey and white matter infarcts. Four relapses: new MR abnormalities | (iii) (iv) (v) | |
| 7 (16F) | MVD | Anton-Babinski syndrome | Normal | Normal cell count, protein | Normal at presen- tation; repeat: occipital infarct | Multiple high signals; cortical gyri, both occipitals; pre-, post-gyri, temporal, thalamus | Occluded central arteries: occipital poles bilaterally: gross luxury perfusion round occipital infarct | (ii) (v) |
| 8 (29F) | MVD | Aphasia, progressive hemiparesis | Normal | Normal | Normal | * | Characteristic vasculitic shoulder: progression on repeat images | (ii) (v) |
| 9 (28M) | MVD | Headache, ipsilateral cerebellar signs, hemiparesis, childhood polymyositis | Normal | Normal | Normal | Infarcts in cerebellum and contralateral internal capsule | Minor peripheral vessel abnormality | (iii) (v) |
| 10 (48M) | MVD | Occipital headache, cerebellar CVA | Normal | Not performed | Evolving left cerebellar infarct | * | Multiple vessels: characteristic vasculitis, extending beyond affected territories on MR | (ii) (iii) (v) |
| 11 (32M) | MVD | Quadriparesis, grand mal seizures | ESR 70, CRP 206 | Normal | Normal | Multiple vessel territory infarcts | Multiple vessels: characteristic vasculitis, | (ii) (iii) (v) |
| 12 (29F) | MVD | Aphasia, hemiplegia | Normal | Normal | Normal | Middle cerebral infarct | Internal carotid: vasculitis, progressing on repeat images | (ii) (iii) (v) |
| Patient (age, sex) | PACNS type | Clinical presentation | Presentation acute phase parameters | CSF | CT | MR* | Angiography* | PACNS diagnostic criteria† |
|---|---|---|---|---|---|---|---|---|
| 1 (55M) | SVD | Confusion, headache, focal seizures, dysphasia | ESR 50, CRP 20 | Protein 0.7 g/l; cell count normal. Negative oligoclonal bands | Cerebral atrophy; dilation ventricles and sulci | Multifocal grey and white matter infarcts: right caudate nucleus, lateral ventricles: Relapses: new abnormalities | Peripheral small vessels hypoperfused | (iii) (iv) (v) |
| 2 (22F) | SVD | Headaches, grand mal seizures | Normal | Normal protein, cell count | Normal | Multiple white and grey matter infarcts* | Abnormal (venous phase) | (ii) (iii) (iv) (v) |
| 3 (32F) | SVD | Hemiparesis, encephalitic | ESR 32, CRP 50 | Normal; negative for oligoclonal bands at presentation and relapse | Normal | Multiple grey and white matter infarcts. Relapses: new MR abnor- mality | Normal | (iii) (iv) (v) |
| 4 (51F) | SVD | Migraines, grand mal seizures | ESR 38, CRP 55 | Protein 0.79 g/l; normal cell count | Acute lacunar infarct, left internal capsule | Multiple grey and white matter infarcts | Normal | (iii) (iv) (v) |
| 5 (53F) | SVD | Encephalitic, grand mal seizures | ESR 80, CRP 40 | Protein (0.74 g/l); WCC 20 × 106/l; 90% lymphocytes; RBC 4 × 106/l | Normal | Pulvinar sign* | (Not performed) | (i) (Brain biopsy) (iv) (v) |
| 6 (27M) | SVD | Lateral medullary syndrome | ESR 30, CRP 30 | Traumatic tap: normal WCC differential/RBC ratio | Normal | Multiple grey and white matter infarcts. Four relapses: new MR abnormalities | (iii) (iv) (v) | |
| 7 (16F) | MVD | Anton-Babinski syndrome | Normal | Normal cell count, protein | Normal at presen- tation; repeat: occipital infarct | Multiple high signals; cortical gyri, both occipitals; pre-, post-gyri, temporal, thalamus | Occluded central arteries: occipital poles bilaterally: gross luxury perfusion round occipital infarct | (ii) (v) |
| 8 (29F) | MVD | Aphasia, progressive hemiparesis | Normal | Normal | Normal | * | Characteristic vasculitic shoulder: progression on repeat images | (ii) (v) |
| 9 (28M) | MVD | Headache, ipsilateral cerebellar signs, hemiparesis, childhood polymyositis | Normal | Normal | Normal | Infarcts in cerebellum and contralateral internal capsule | Minor peripheral vessel abnormality | (iii) (v) |
| 10 (48M) | MVD | Occipital headache, cerebellar CVA | Normal | Not performed | Evolving left cerebellar infarct | * | Multiple vessels: characteristic vasculitis, extending beyond affected territories on MR | (ii) (iii) (v) |
| 11 (32M) | MVD | Quadriparesis, grand mal seizures | ESR 70, CRP 206 | Normal | Normal | Multiple vessel territory infarcts | Multiple vessels: characteristic vasculitis, | (ii) (iii) (v) |
| 12 (29F) | MVD | Aphasia, hemiplegia | Normal | Normal | Normal | Middle cerebral infarct | Internal carotid: vasculitis, progressing on repeat images | (ii) (iii) (v) |
ESR, erythrocyte sedimention rate (mm/h); CRP, C-reactive protein (mg/l). *Findings displayed in Figures 7–10. †Diagnostic criteria for PACNS: (i) brain biopsy; (ii) characteristic angiographic findings; (iii) multiple CNS artery territory white and grey matter infarcts temporally related; (iv) relapsing course with new infarcts found on MR; (v) clinical response(s) to immunosuppressive regimen.
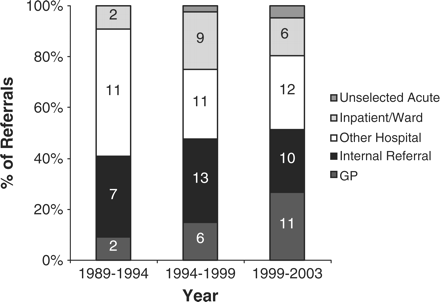
Trends in referral source pattern for all vasculitides, showing significant increases from primary care (p = 0.01).
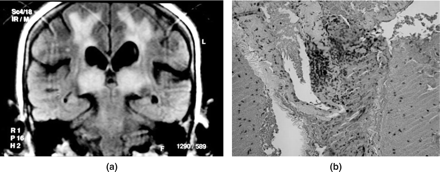
(a) The pulvinar sign, patient 5, coronal flair image: generally considered diagnostic of new variant Creutzfeldt-Jakob disease, presenting in acute small-vessel PACNS encephalopathy in a 53-year-old female. White matter ischaemic changes in both hemispheres are also present. (b) Brain biopsy from patient 5, showing fibrinoid necrosis with small-vessel vasculitis.
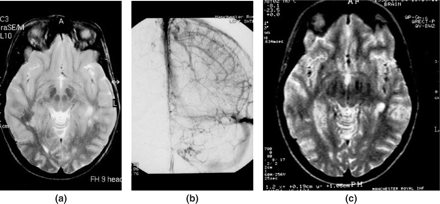
Patient 2, a 22 year old female receiving the oral contraceptive pill, presenting with grand mal convulsions due to small-vessel PACNS. (a) MR scan, axial T2-weighted tubular spin echo (TSE) showing diffuse grey and white matter ischaemic lesions. (b) Direct cerebral angiography showing no evidence of vascular occlusions, but contrast pooling, capillary blush apparent during the venous phase. (c) MR scan, during relapse 4 years later, with widespread severe ischaemic injuries.
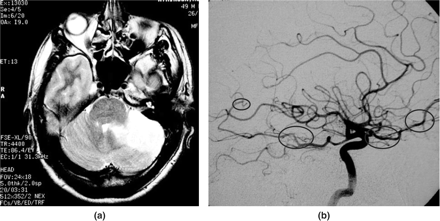
Patient 10, a 49 year old male, presenting with cerebellar infarct due to middle-sized vessel PACNS. (a) MR T2-weighted fast spin echo showing left cerebellar signal change only. (b) Direct cerebral angiography filling only of the right posterior inferior cerebellar artery, left absent; narrowing of the distal basilar artery due to vasculitis; other abnormalities encircled. The cerebral angiogram revealed multiple vessels showing characteristic MVD vasculitis, several without MR scan abnormalities in corresponding territories.
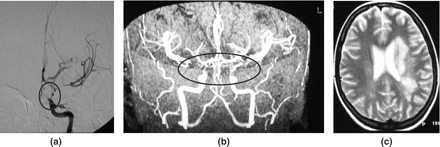
Patient 8, a 26 year old female, with hemiparesis and aphasia due to middle-sized vessel PACNS. (a) Selective left internal carotid angiography: severe stenosis of the distal internal carotid artery, proximal anterior and middle cerebral arteries, progressing 7 days later with similar lesions on the right. (b) MR angiography. (c) Limited left hemisphere abnormalities on T2-weighted MR scan.
Referral source: primary and secondary care, all vasculitides
We analysed individual referral rates for each subgroup over each of the 5-year periods, using Poisson regression analysis, for the 97 patients whose referral patterns were documented. There was a significant increase in the rate of referrals from primary care (p = 0.01), but the other sources showed no change (Figure 6).
Discussion
Our results show increase in the overall frequency of presentation of all systemic vasculitides during the past 15 years. The PRR of 1.323 is consistent with studies by Watts,15 where they estimated an incidence of 19.8 per million in 2000, compared with an earlier estimate of 10 per million in 1982 by Scott.2 Such measurements do not necessarily correspond to an increase in incidence—there may have been heightened diagnostic awareness as ACR and CHCC criteria became established and broadly implemented. Likewise, our data, with potential variation in the population denominator as more centres became established regionally, do not prove incidences are rising. However the stability of presentation numbers for most of the vasculitides, contrasting with striking rises in PAN and PACNS, suggest that there may have been a true increase in incidence of both the latter diseases.
Our patients with multi-system vasculitis were younger than those in other studies, with a mean age of 46.2 years (median 46.5), contrasting with (e.g.) that found by Watts:15 mean age with systemic vasculitis, 62.9 years, median 65 years, although their sample group did not include PACNS, HSP, secondary SLE, or cutaneous polyarteritis. Carruthers16 suggested that older patients were less likely to be referred to tertiary care, but against this, clinical features seem similar in patients in tertiary care compared to those in secondary care. Previous studies have suggested that systemic vasculitis is more common in men,15,17,18 but our data this did not confirm this, although there was a male bias for WG, CSS, MPA and PAN.
PACNS, both SVD and MVD, presented only in the 6 years since 1998, with a total of 12 patients, mean age 36 years. The PRR of 5.605 represents a highly significant increase. We interpret these findings as favouring a true increase in incidence for three reasons: the relative number of cases compared to the multi-system diseases; the consistency of interests, referral patterns, and imaging techniques of the physicians and radiologists in the neurosciences and systemic vasculitis fields, and the earlier documentation of multiple cases of secondary CNS vasculitis (Figure 4) from 1994 to 1998, in the absence of PACNS cases.
Delineation of the natural histories of relapses in PACNS of SVD and MVD types showed distinct clinical courses. SVD PACNS relapsed in 30% in the first 2 years, a figure remarkably similar19 to that of its putative systemic vasculitis equivalent, MPA, using a widely-used treatment regimen for the latter, reinforcing the view that SVD PACNS represents a limited form of MPA. The continued relapses in SVD PACNS at a similar rate, to 6 years of follow-up is of concern, because of consequential irreversible CNS injury, occurring in half of such patients during the first 2 years during treatment, while they were receiving low-dose prednisolone and azathioprine. Relapses occurring 2 or more years after initial presentation occurred while patients were no longer receiving immunosuppressives. All relapses in SVD PACNS were successfully re-induced into clinical remission after further courses of initial induction therapy (prednisolone with oral cyclophosphamide, 1.5 mg/kg/day for 3 months), but all patients had suffered further neurological injury both clinically and radiologically, on MR scanning (Figure 8).
Diagnosis and management of SVD PACNS is more problematic than that of the MVD variant. Direct angiography of the cerebral circulation in SVD proved unhelpful in establishing a diagnosis, all angiograms showing no evidence of occlusions at the limits of resolution of the technique, although one patient showed some evidence of capillary pooling of contrast in the venous phase (Figure 8). This conclusion, together with our finding of the pulvinar sign (considered diagnostic of nvCJD,20 with high specificity and sensitivity for this disease), in patient 5, where brain biopsy confirmed SVD PACNS (Figure 7), makes it essential to maintain a high index of suspicion for this form of PACNS, both in presentation and in relapse. Although brain biopsy, if found to be positive, is the standard for diagnosis of small-vessel cerebral vasculitis, its risks are significant, with mortality reported as 3.3%, comparable with risks of serious side-effects from long-term immunosuppressive therapy. Risks associated with the procedure may be reflected in the finding that up to 75% of reported cases are diagnosed without histopathology.21 Our data also support the idea that brain biopsy should not be a sine qua non for PACNS, with a false negative rate of 30% reported in patients with an otherwise unequivocal diagnosis.22 Advocates of early investigation by brain biopsy may not have wholly incorporated these issues in considerations for safest effective patient management.23–25
Findings in our series of SVD PACNS, and knowledge of its natural history, also strongly support the case for alternative maintenance immunosuppressives, extending at least to 5 years after presentation: these might include more prolonged use of corticosteroids with cyclophosphamide at low dose, although the efficacy of this regimen in preventing relapses has been placed in doubt for systemic small vessel vasculitides,19 or alternatively, with newer agents having both T- and B-cell effects, such as the mycophenolate derivatives.
In contrast, middle-size vessel PACNS showed angiographic abnormalities in all cases, with distinct, multiple-vessel territories a hallmark. MR scans did not necessarily reflect the extent of angiitis demonstrable on angiography (Figures 9 and 10). The use of MR scanning in conjunction with cerebral angiography in cases with a high index of clinical suspicion has become an essential in establishing the diagnosis. Imbesi9 described a case where the MR scan was negative despite angiography showing evidence of foci of irregular narrowing within the cerebral vasculature. This inconsistency was thought to be due to timing, the MR scan not being positive during the process of ischaemic change. They concluded that this showed the necessity to perform angiography in all cases with high clinical suspicion. Our findings show that normal areas of the CNS on MR scan can coexist temporally with abnormalities on angiography in MVD PACNS, presumably because ischaemia has not yet become critical. This emphasizes the need for angiography in all cases where there is a high index of suspicion of middle-size vessel vasculitis. Demonstration of concurrent multiple-vessel territory disease is an uneqivocal diagnostic criterion, as is characteristic single vessel abnormality in patients who do not have evidence of atheromatous disease, both of higher diagnostic sensitivity than MR scan alone. Our findings suggest an optimal risk–benefit diagnostic algorithm for patients when SVD or MVD PACNS is suspected (Figure 11).
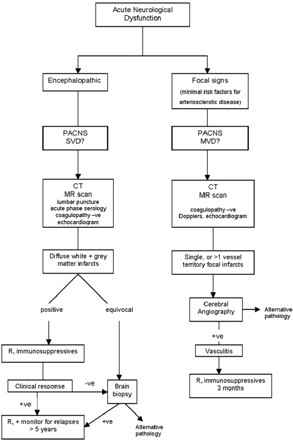
Algorithm for management of suspected small-vessel, or middle-sized vessel PACNS, suggested from analysis of this series.
All patients with MVD PACNS were relapse-free during 6 years follow-up, and all had discontinued low-dose prednisolone and azathioprine by 24 months from presentation. Its potential systemic equivalent PAN, both in our experience (data not shown), and that of others,26 (both systemic and the cutaneous-limited variant), is an indolent, frequently relapsing vasculitis, with a propensity to cause progressive organ injuries over long periods: MVD PACNS is more benign after initial presentation than either PAN or PACNS SVD.
The natural histories of SVD and MVD PACNS are strikingly distinct, as are their clinical, serological acute-phase, and radiographic characteristics. Our findings suggest that PACNS is not a unifying diagnosis, and that these distinctions should be considered when planning treatment for the two variants of PACNS. Although acute-phase parameters are useful in designating inflammatory processes at presentation in SVD PACNS, they are unhelpful in defining relapses in this form of PACNS, the diagnosis of which rested in all cases on clinical acumen and MR scans. Delineation of the two forms of PACNS has important implications in optimizing risks and benefits of immunosuppressive regimens. In SVD PACNS, treatment should be at least as intensive, and potentially more prolonged than for the equivalent antineutrophil antibody-positive small-vessel systemic vasculitis, whereas in MVD PACNS, our data suggest that immunosuppressives can safely be discontinued after 2 years or less. SVD and MVD PACNS may be best considered separate, frequently encountered, entities in the context of secondary and tertiary referral practice in systemic vasculitis.
References
Scott DG, Bacon PA, Elliott PJ, Tribe CR, Wallington TB. Systemic vasculitis in a district general hospital 1972–1980: clinical and laboratory features, classification and prognosis of 80 cases.
Calabrese LH, Mallek JA. Primary angiitis of the CNS: report of 8 new cases, review of the literature, and proposal for diagnostic criteria.
Crane R, Dublin Kerr L, Spiera H. Clinical analysis of isolated angiitis of the central nervous system.
Wasserman BA, Stone JH, Hellmann DB, Pomper MG. Reliability of normal findings on MR imaging for excluding the diagnosis of vasculitis of the central nervous system.
The American College of Rheumatology 1990 Criteria for the Classification of Wegener's Granulomatosis.
Jennette JC, Falk EJ. Nomenclature and Classification of Vasculitis: Proposal of International Consensus Conference.
Calabrese LH, Duna GF, Lie JT. Vasculitis in the Central Nervous System.
Imbesi SG. Diffuse Cerebral Vasculitis with Normal Results on MR Imaging.
Calabrese LH, Duna GF. Evaluation and Treatment of Central Nervous System Vasculitis.
Fauci AS, Haynes B, Katz P. The spectrum of vasculitis: clinical, pathologic, immunologic and therapeutic considerations.
Scolding NJ, Jayne DRW, Zajicek JP, Meyer PAR, Wraight EP, Lockwood CM. Cerebral Vasculitis—recognition, diagnosis and management.
Bruce IN, Bell AL. A Comparison of two Nomenclature Systems for Primary Systemic Vasculitis.
Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of Systemic Vasculitis. A Ten-Year Study in the United Kingdom.
Carruthers DM, Watts RA, Symmons DPM, Scott DG. Wegner's Granulomatosis—increased incidence or increased recognition?
Watts RA, Gondalez-Gay MA, Lane SE, Garcia-Porrua SE, Benthan G, Scott DGI. Geoepidemiology of systemic vasculitis: comparison of the incidence in two regions of Europe.
Watts RA, Carruthers DM, Scott DGI, Epidemiology of Systemic Vasculitis—Changing Incidence or Definition?
Jayne D, Rasmussen N, Andrassy K, et al. A Randomized Trial of Maintenance Therapy for Vasculitis associated with Antineutrophil Cytoplasmic Antibodies.
Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M, Knight R, Will RG. Diagnosing Variant Creutzfeldt-Jakob Disease with the Pulvinar Sign: MR Imaging Findings in 86 Neuropathologically Confirmed Cases.
Lie JT. Classification and Histopathological Spectrum of Central Nervous Sytem Vasculitis.
Alrawi A, Trobe JD, Blaivas M, Musch DC. Brain Biopsy in the Diagnosis of Primary Angiitis of the Central Nervous System.
Lanthier S, Lortie A, Michaud J, Laxer R, de Veber G. Isolated angiitis of the CNS in Children.
Volcy M, Toro ME, Uribe CS, Toro G. Primary angiitis of the central nervous system: report of five biopsy-confirmed cases from Colombia.
Author notes
Departments of General Medicine, 1Radiology, and 2Neurology, Manchester Royal Infirmary, Manchester, UK

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}