





Abstract
Pleomorphic xanthoastrocytoma (PXA) is a rare astrocytic neoplasm of the brain. Some PXAs are accompanied by anaplastic features and are difficult to manage because of frequent recurrences that lead to early death. No previous reports have demonstrated consistent efficacy of adjuvant radiotherapy or chemotherapy for this disease. We report a case of PXA with anaplastic features treated with stereotactic irradiation (STI) that resulted in long-term control of repeatedly recurring nodules throughout the neuraxis. A 47-year-old woman presented with an epileptic seizure due to a large tumor in the right frontal lobe. The tumor was resected and diagnosed as PXA with anaplastic features. Sixteen months later, a relapse at the primary site was noted and treated with stereotactic radiosurgery using Gamma Knife. Two years later, the patient developed a tumor nodule in the cervical spinal cord that histologically corresponded to a small-cell glioma with high cellularity and prominent MIB-1 (mindbomb homolog 1) labeling. In the following months, multiple nodular lesions appeared throughout the CNS, and STI was performed six times for eight intracranial lesions using Gamma Knife and twice using a linear accelerator, for three spinal cord lesions in total. All lesions treated with STI were well controlled, and the patient was free from symptomatic progression for 50 months. However, diffuse dissemination along the craniospinal axis eventually progressed, and she died 66 months after initial diagnosis. Autopsy showed that the nodules remained well demarcated from the surrounding nervous system tissue. STI may be an effective therapeutic tool for controlling nodular dissemination of PXA with anaplastic features.
Pleomorphic xanthoastrocytoma (PXA) is a rare tumor accounting for less than 1% of all astrocytic neoplasms.1–4 Maximum surgical removal is considered the first treatment of choice.1 The efficacy of adjuvant radiotherapy or chemotherapy has not yet been established, largely because of the relative rarity of this disease.2,5–7 PXA belongs to grade II of the WHO histological classification of tumors of the CNS and is therefore considered as a relatively benign entity among the astrocytic tumors.4,8,9 However, 9%–20% of PXAs have been reported to undergo malignant transformation,10 and some of them exhibit anaplastic features at the first presentation.11 PXAs with anaplastic features are difficult to manage, and the role of postoperative radiotherapy is not clearly defined.2,5,12,13 We present a case of PXA with anaplastic features in which we controlled disseminated tumor nodules in the brain and the spinal cord for a relatively long period by repeated stereotactic irradiation (STI).
Case History
A 47-year-old woman presented with an epileptic seizure. CT scanning of the head revealed a 6-cm tumor in the right frontal lobe. MRI showed a well-demarcated, homogeneously enhanced mass lesion accompanied by perifocal edema and midline shift (Fig. 1). The tumor was totally removed, and histological examination revealed a tumor primarily composed of spindle cells with fibrillary cytoplasm (Fig. 2A), arranged in a fascicular pattern. Round or polygonal cells with plumper cytoplasm were admixed, and large pleomorphic cells and multinucleated giant cells were scattered throughout the lesion (Fig. 2B). Reticulin fibers were observed around individual or grouped tumor cells (Fig. 2C). Focally, eosinophilic granular bodies were noted (Fig. 2D). On immunohistochemical examination, tumor cells in some areas were reactive for glial fibrillary acidic protein (Fig. 2E). Cellularity was high, and several mitotic figures were frequently observed. MIB-1 (mindbomb homolog 1) labeling index was 4% (Fig. 2F). These findings indicated that the tumor was an astrocytic glioma with signs of anaplasia, including high cellularity, increased cellular and nuclear atypia, and focally increased mitotic activity. Histopathological features highly indicative of PXA included cellular pleomorphism, focal eosinophilic protein droplets, and regions with an interstitial reticulin fiber network. A rather solid, noninfiltrative growth of the tumor was also typical with PXA. Furthermore, molecular analysis of the tumor revealed two distinct mutations in exon 4: a missense mutation resulting in an amino acid change at codon 114 (c. 341T>C; L114S) and a nonsense mutation due to a 2-bp deletion (c. 365–366delTG). Both mutations do not correspond to any of the known hotspot mutations typically found in diffuse astrocytic gliomas.14 Alterations of the EGFR (epidermal growth factor receptor), CDK4 (cyclin-dependent kinase 4), MDM2 (murine double minute 2), and CDKN2A (cyclin-dependent kinase inhibitor 2A) genes were not observed, in keeping with what has been reported in the majority of PXAs.15–17 The histopathological findings together with these molecular analysis results led to the diagnosis of PXA with anaplastic features. The pathological diagnosis was independently confirmed by three neuropathologists.
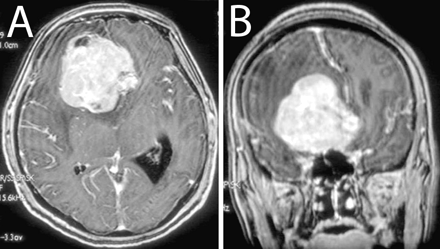
Axial (A) and coronal (B) gadolinium-enhanced T1-weighted MRI at presentation.
Sixteen months after the first operation, a focal relapse at the primary site was noted and treated with stereotactic radiosurgery (SRS) using Gamma Knife by irradiating 20 Gy at the margin of the tumor (Fig. 3A). Two years later, surgical removal with laminectomy was performed on intrathecal dissemination at the level of the fourth to fifth cervical spine. Histology of this recurrent tumor was analogous to that of the original tumor. However, the recurrent tumor was largely composed of small cells (Fig. 2G), and the MIB-1 labeling index had increased to 10% (Fig. 2H).
At 40 months after the original diagnosis, a new intracranial lesion appeared in the left hypothalamus. Nimustine hydrochloride (100 mg/body) was administered intravenously twice in 2 months, but the lesion showed a poor response to chemotherapy and continued to grow. This lesion was therefore treated by the second SRS using Gamma Knife at 43 months (Fig. 3B). Relapses of several nodular lesions apart from the primary tumor were observed. Spinal cord lesions at the levels of the fourth cervical spine and the fourth thoracic spine were treated with STI at 47 months (Fig. 4A). For this STI, treatment planning with irregularly shaped beams using seven noncoplanar static ports was established with the help of a three-dimensional treatment-planning computer, and the patient was immobilized using a stereotactic body frame. Forty-two Gy in 12 fractions was administered with 6-MV X-rays using a linear accelerator. Newly disseminated lesions in the right temporal lobe and the left cerebellomedullary angle were treated with a third SRS at 50 months (Fig. 3C, D). One month after the third SRS, a spinal cord lesion at the level of the 10th to 12th thoracic spine was partially resected followed by STI delivering 40 Gy in 10 fractions. One dynamic arc instead of multiple static ports was employed for the second STI. At 53 months, temozolomide was administered orally at a dose of 150 mg/m2 using the 5/28-day protocol, and the patient received six cycles in total. At 55 and 58 months, a fourth and fifth SRS were performed for the lesions in bilateral cerebellar hemispheres (Fig. 3E, F) and the frontal interhemispheric region (Fig. 3G), respectively. At 59 months, the spinal cord lesion at the level of the first to third cervical spine received 35 Gy irradiation in 10 fractions, and another new lesion in the left temporal lobe was treated with a sixth SRS (Fig. 3H).
All of the intracranial lesions treated with SRS and the spinal cord lesions that received STI had successful local control (Figs. 4B, 5A–H), and no obvious clinical complications occurred from STI. The patient was free from neurological deficits until the thoracic spinal cord lesion started to cause paraparesis and bladder and bowel dysfunction at 50 months. However, at 60 months, numerous tumor nodules appeared simultaneously throughout the craniospinal axis, which was beyond control by STI. Consciousness deteriorated due to rapid progression of the diffuse dissemination, and she died 66 months after the disease onset.
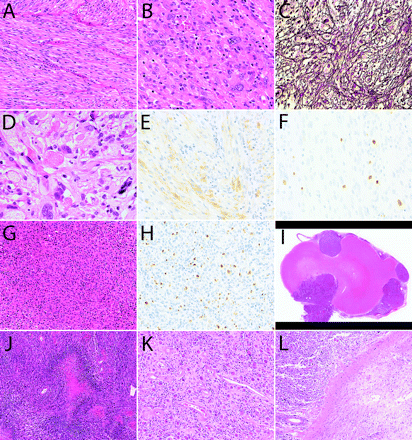
(A–F) First surgical specimen demonstrating spindle cells (A), pleomorphic cells (B), reticulin fibers (C), eosinophilic protein droplets (D), and cells positive for glial fibrillary acidic protein (E). MIB-1 (mindbomb homolog 1) labeling index was 4% (F). (G and H) Recurrent tumor showing high cellularity (G) and MIB-1 labeling index (H). (I–L) Autopsy specimens showing well-demarcated (I), highly cellular tumor (J) and less cellular region (K) adjacent to degenerated white matter (L). Staining: A, B, D, G, and I–L, hematoxylin and eosin staining; C, silver staining; E, immunostaining for glial fibrillary acidic protein; F and H, MIB-1 staining. Original magnification: A, G, and K, ×200; B, C, E, F, and H, ×400; D, ×800; I, ×10; J and L, ×100.
Autopsy demonstrated multiple disseminated lesions throughout the craniospinal axis. We could not identify the primary lesion. Each disseminated tumor nodule appeared relatively well demarcated from the surrounding brain tissue, although there was marked associated edema (Fig. 2I). Histologically, the specimens were highly cellular malignant astrocytic tumors with multifocal areas of necrosis and pseudopalisading (Fig. 2J). The tumors grew diffusely within the leptomeninges and in the Virchow-Robin spaces along intracortical vessels. Features characteristic of PXA were no longer present in the disseminated lesions. The lesions treated by SRS were specifically examined. Macroscopically, the irradiated tumors had shrunk and were difficult to recognize. Histological evaluation of these irradiated tumors showed areas with lower cellularity (Fig. 2K), but some areas remained highly cellular, similar to untreated tumors. Irradiated tumors were typically adjacent to degenerated white matter, which probably reflected damage from irradiation (Fig. 2L). In the spinal cord, radionecrotic spots were observed in regions adjacent to the STI-treated lesions (Fig. 4C).
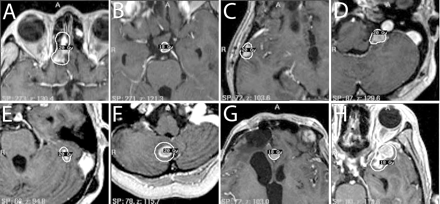
Treatment planning for the first (A), second (B), third (C and D), fourth (E and F), fifth (G), and sixth (H) SRS.
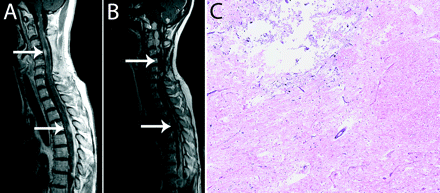
(A and B) Gadolinium-enhanced T1-weighted MRI of the spinal cord showing lesions before STI (A, white arrows) that were controlled until 60 months (B, white arrows). (C) Autopsy specimen of the spinal cord adjacent to irradiation field showing the area of radiation necrosis (hematoxylin and eosin staining; original magnification, ×50).
Discussion
Primarily because of the rarity of PXA with anaplastic features,11,18 the role of standard postoperative therapy for PXA with anaplastic features has not been definitively established. Seventeen cases of PXA showing anaplastic features at the first presentation have been reported.19 Among 13 patients whose follow-up information was available, five patients died, and the average overall survival was 21 months (range, 1–44 months). For the other eight patients, the last follow-up was at 43 months average (range, 3–144 months).19 Nine of 17 patients underwent conventional fractionated radiotherapy, but a therapeutic benefit has not been apparent.19 Notably, in the present case, STI was efficacious in controlling the disseminated nodules of PXA with anaplastic features up to a certain point of progression. By the use of repetitive STI, the patient was free from neurological deficit for 50 months despite frequent tumor recurrences within a short duration. Leptomeningeal dissemination is not an uncommon pattern of recurrence for PXA with anaplastic features and has been observed in two of six recurrent cases.19 Leptomeningeal dissemination of PXA, with or without anaplastic features, commonly presents as multiple nodular lesions, in contrast to the diffuse dissemination seen in high-grade astrocytomas.20 Consistently, in the present case, the dissemination caused numerous nodular lesions, but diffuse enhancement of the subarachnoid space was not seen until the last few months. Moreover, at autopsy, the remaining nodules were demonstrated to be well demarcated from the surrounding brain and spinal cord. Such nodular lesions would be amenable to local therapy. In fact, the efficacy of STI for controlling the disseminated nodules was observed by both MRI and histological examinations. In addition, multiple STI treatments were well tolerated, including those to spinal cord lesions, and no complications were noted, although degeneration of adjacent neural tissue was found at autopsy. The use of STI for intramedullary spinal cord malignant tumors has not been reported; however, CyberKnife radiosurgery has been used for spinal benign tumors and arteriovenous malformations and has demonstrated low morbidity.21,22
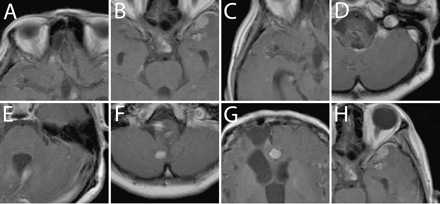
Gadolinium-enhanced T1-weighted MRI at 60 months. A–H correspond to the lesions treated with SRS shown in Fig. 3A–H.
PXA with anaplastic features can show transformation to glioblastoma7,23 as in the present case; the histology of the disseminated nodules eventually mimicked that of glioblastoma. PXA with anaplastic features at diagnosis typically has a poor prognosis.11,24 In the present case, the relatively long-term control was obtained by use of STI. The relationship of such control to delaying frankly malignant progression to glioblastoma remains conjectural.
Chemotherapy for PXA has been generally considered ineffective,2,6 although a case has been reported in which the use of vincristine and carboplatin was effective for controlling bleeding during surgery, presumably due to devascularization.25 We administered nimustine hydrochloride, according to the regimen for high-grade astrocytoma, and temozolomide, after it became available, but whether these chemotherapeutic reagents contributed to the relatively long survival of the patient is unknown. Rather, the repeated appearance of disseminated nodules during the course of nimustine hydrochloride treatment suggests that the drug was ineffective. Temozolomide was administered only in the late stage of progression, and whether upfront administration of temozolomide is useful awaits further cases; the use of temozolomide for this disease has not been reported previously. Clearly, there is a need for systemic therapy because repeated STI did not cure the disease.
In summary, we present a case of PXA with anaplastic features in which repeated local and distant recurrences were controlled by means of STI for a relatively long period. Whereas conventional chemotherapy and radiotherapy are not curative for PXA with anaplastic features, STI may be a useful therapeutic tool for controlling nodular disseminations and improving quality of life in patients with this disease.
We acknowledge Drs. Masahiro Shin and Masao Tago for their participation in radiation therapy.
References
Fouladi M, Jenkins J, Burger P, et al. Pleomorphic xanthoastrocytoma: favorable outcome after complete surgical resection.
Giannini C, Scheithauer BW, Burger PC, et al. Pleomorphic xanthoastrocytoma: what do we really know about it?
Gomez JG, Garcia JH, Colon LE. A variant of cerebral glioma called pleomorphic xanthoastrocytoma: case report.
Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases.
Cervoni L, Salvati M, Santoro A, Celli P. Pleomorphic xanthoastrocytoma: some observations.
Thomas C, Golden B. Pleomorphic xanthoastrocytoma: report of two cases and brief review of the literature.
Whittle IR, Gordon A, Misra BK, Shaw JF, Steers AJ. Pleomorphic xanthoastrocytoma. Report of four cases.
Stuart G, Appleton DB, Cooke R. Pleomorphic xanthoastrocytoma: report of two cases.
Giannini CPW, Louis DN, Liberski P. Pleomorphic xanthoastrocytoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds.
Chakrabarty A, Mitchell P, Bridges LR, Franks AJ. Malignant transformation in pleomorphic xanthoastrocytoma—a report of two cases.
Lubansu A, Rorive S, David P, et al. Cerebral anaplastic pleomorphic xanthoastrocytoma with meningeal dissemination at first presentation.
Bayindir C, Balak N, Karasu A, Kasaroglu D. Anaplastic pleomorphic xanthoastrocytoma.
Bucciero A, De Caro MI, Tedeschi E, et al. Atypical pleomorphic xanthoastrocytoma.
Koga H, Zhang S, Kumanishi T, et al. Analysis of p53 gene mutations in low- and high-grade astrocytomas by polymerase chain reaction-assisted single-strand conformation polymorphism and immunohistochemistry.
Weber RG, Hoischen A, Ehrler M, et al. Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas.
Kaulich K, Blaschke B, Numann A, et al. Genetic alterations commonly found in diffusely infiltrating cerebral gliomas are rare or absent in pleomorphic xanthoastrocytomas.
Paulus W, Lisle DK, Tonn JC, et al. Molecular genetic alterations in pleomorphic xanthoastrocytoma.
Hirose T, Ishizawa K, Sugiyama K, Kageji T, Ueki K, Kannuki S. Pleomorphic xanthoastrocytoma: a comparative pathological study between conventional and anaplastic types.
Marton E, Feletti A, Orvieto E, Longatti P. Malignant progression in pleomorphic xanthoastrocytoma: personal experience and review of the literature.
Nakajima T, Kumabe T, Shamoto H, Watanabe M, Suzuki H, Tominaga T. Malignant transformation of pleomorphic xanthoastrocytoma.
Dodd RL, Ryu MR, Kamnerdsupaphon P, Gibbs IC, Chang SD Jr, Adler JR Jr. CyberKnife radiosurgery for benign intradural extramedullary spinal tumors.
Sinclair J, Chang SD, Gibbs IC, Adler JR Jr. Multisession CyberKnife radiosurgery for intramedullary spinal cord arteriovenous malformations.
Kepes JJ, Rubinstein LJ, Ansbacher L, Schreiber DJ. Histopathological features of recurrent pleomorphic xanthoastrocytomas: further corroboration of the glial nature of this neoplasm. A study of 3 cases.
Hirose T, Giannini C, Scheithauer BW. Ultrastructural features of pleomorphic xanthoastrocytoma: a comparative study with glioblastoma multiforme.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}