Article Text

Abstract
BACKGROUND Malondialdehyde (MDA) in plasma is regarded as an indicator for increased lipid peroxidation.
METHOD Measurements of MDA concentrations in plasma were compared among healthy children (n=31), patients with neurological disorders or epileptic syndromes (n=15), and children with pontocerebellar structural defects (n=31), where the cause or genetic defect remained unknown.
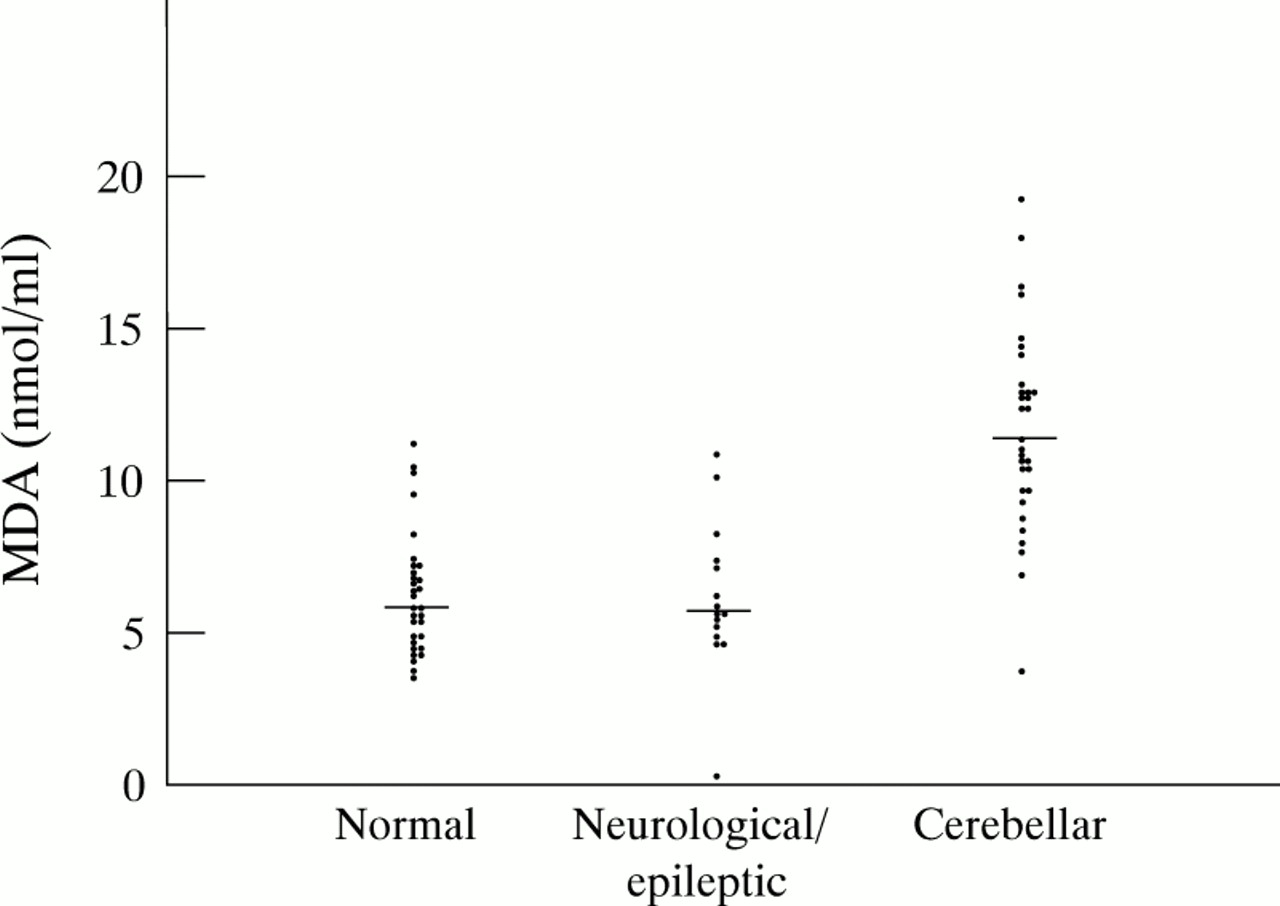
RESULTS In healthy children the median MDA value was 5.86 nmol/ml (mean (SD) value: 6.25 (1.97), range: 3.76–11.19). For the group with various neurological disorders or epilepsy, the values were similar with the median value at 5.66 nmol/ml (range 0.22–10.86). Compared with healthy controls and the neurological/epileptic group, the 31 children with pontocerebellar structural defects had significantly increased MDA values with a median value at 11.29 nmol/ml (mean (SD) value: 11.62 (3.27), range 3.65–19.22).
IMPLICATION These findings of increased plasma MDA in the majority of children with pontocerebellar structural defects of unknown origin raised the question whether increased lipid peroxidation leads to prenatal and postnatal pontocerebellar maldevelopment or degeneration.
- malondialdehyde
- lipid peroxidation
- cerebellum
Statistics from Altmetric.com
Oxidative stress is defined as oxygen radical mediated damage to biological material (proteins, lipids, carbohydrates, and DNA) caused by either increased generation and build up of the oxygen radicals (superoxide radical, hydrogen peroxide, and the hydroxyl radical), or due to the diminished removal or inadequate protection against these ubiquitously present oxygen radicals. During evolution of aerobic life, powerful antioxidant enzymes and radical scavengers have developed to remove or scavenge these oxygen radicals in order to protect cell organelles and membranes.1-4
In experiments on cerebellar development in the mouse, gamma irradiation and several toxins are known exogenous agents to generate increased free radicals setting off a mechanism of programmed cell death or apoptosis in cerebellar granule cells. Early apoptosis induced by these exogenous factors results in cerebellar hypoplasia, as cerebellar granule cells are responsible for the bulk of volume growth in the developing cerebellum.5 Increased generation of reactive oxygen species and lipid peroxidation followed by apoptosis has also been shown recently in cultures of cortical neurons from fetal brains with Down’s syndrome. Degeneration of Down’s syndrome neurons could be prevented by treatment with free radical scavengers or catalase.6 The known moderate cerebellar hypoplasia in patients with Down’s syndrome might reflect the effect of early degeneration of cerebellar progenitor cells by increased lipid peroxidation and apoptosis in vivo. Progressive cerebellar hypoplasia or atrophy has also been found to be a prominent finding in some well defined mitochondrial diseases of the central nervous system where increased oxidative stress might play an important part in the pathogenesis of these disorders.2 4 7 8 Examples of these conditions included mitochondrial diseases such as Kearns-Sayre syndrome, myoclonic epilepsy with ragged red fibres (MERRF) syndrome, and cytochrome c oxidase deficiency. Owing to the block of electron flow alongside the respiratory chain enzymes, electrons will leak and produce more superoxide radicals. The consequent production of hydroxyl radical and other powerful radicals can initiate a chain reaction of lipid peroxidation in which polyunsaturated fatty acid side chains are converted into lipid peroxides. One of the end products of lipid peroxidation is malondialdehyde (MDA).
Based on these observations, we decided to screen patients with pontocerebellar maldevelopment or degeneration for MDA as an indicator of increased lipid peroxidation.
Patients and methods
DESCRIPTION OF PATIENTS
A total of 31 healthy children without a previous history of any birth trauma, neurological disorder, or epilepsy served as a reference group for plasma MDA values. The median, mean value, and SD have been calculated. The median age was 7.2 years with a range between 1–19.
MDA values were also determined in a second group of 15 children who were known to suffer from various neurological disorders or epileptic syndromes and in whom neuroimaging had excluded infratentorial structural abnormalities. Their median age was 3 years with a range between 0.8–12 years. In all children, extensive investigations excluded inborn errors of metabolism, mitochondrial, lysosomal, or peroxysomal disorders. Depending on whether frequent daily fits occurred or seizures were only rare events, a further classification distinguished between eight children with epilepsy and seven non-epileptic children. In the group of eight epileptic children, the following clinical entities have been diagnosed: infantile epileptic syndromes with neurodevelopmental retardation in five children, progressive neuronal degeneration with liver disease (Alpers’ disease) in one infant, and occasional fits in two children. In the group of seven non-epileptic children, four children had suffered from seizures or epilepsy in the past but all showed an excellent response to anticonvulsant drug treatment. The latter group included congenital hemiplegia and pseudo-Lennox syndrome in one child, Landau-Kleffner syndrome and electrical status epilepticus during slow sleep in three children, mental retardation with choreoathethosis in two children, and short bowel syndrome after jejunoileal resection for volvulus in one child.
The third group of 31 patients displayed structural pontocerebellar abnormalities of heterogeneous aetiology detected by neuroimaging. Their median age was 8 years with a range from 0.6–20 years. Further classification of this group was made depending on the site of the structural abnormality and on the presence of a static or progressive clinical course. Seven patients suffered from psychomotor retardation associated with unilateral cerebellar hypoplasia.
Five patients suffered from midline fusion defects or vermis agenesis. Among the children with vermis agenesis two patients presented a typical history and clinical findings consistent with Joubert’s syndrome. The three other children had mental retardation with complete or incomplete vermis fusion defects. In one of the children with incomplete vermis fusion defects, the muscle biopsy specimen was consistent with cap myopathy. Non-progressive cerebellar hypoplasia was detected in three children. One child suffered from the acrocallosal syndrome, one from Dandy-Walker syndrome, and one from the Dandy-Walker variant.
Six children had a syndrome with pontocerebellar hypoplasia or atrophy. One child suffered and died from autosomal recessive pontocerebellar hypoplasia type I, three children from pontocerebellar hypoplasia type II, one child suffered from progressive encephalopathy with hypsarrhythmia and optic atrophy (PEHO), and one from progressive microcephaly with pontocerebellar atrophy.
Ten patients suffered from syndromes associated with progressive degeneration of all cerebellar structures. The established diagnosis included the following disease entities: infantile and late infantile type of neuronal ceroid lipofuscinosis, Sjögren-Larsson syndrome, ataxia telangiectasia, Boucher-Neuhäuser syndrome, two siblings with a progressive spastic ataxic syndrome, one patient with an early onset spastic ataxic syndrome with cerebellar atrophy and dysmyelination, and two patients with progressive ataxia associated with a severe polyneuropathy. Except for sporadic fits in the patient with PEHO syndrome and the two patients with infantile and late infantile ceroid lipofuscinosis, all other patients with cerebellar structural defects did not suffer from convulsions.
Table 1 lists an overview of the clinical or descriptive diagnosis. In most disease entities the genetic or neurometabolic disturbance has not yet been identified.
The diagnosis is listed for the different groups of neurological patients without (n=15) and with pontocerebellar involvement (n=31)
MEASUREMENT OF MDA
MDA was measured using the thiobarbituric acid based procedure described by Wasowicz et al.9 Briefly, 50 μl of plasma are added to 0.95 bidistilled water followed by the addition of 1 ml of a solution containing 29 mmol/l thiobarbituric acid in 8.75 mol/l acetic acid. Samples are heated for 60 minutes at 95°C and cooled down, then 25 μl of a solution containing 5 mol/l hydrochloric acid is added.
After adding of 3 ml 1-butanol, samples are centrifuged (1000 ×g, 10 minutes, 20°C) and fluorescence measured (excitation wavelength: 525 nm; emission wavelength: 547 nm).
STATISTICAL ANALYSIS AND SD VALUES
For each group the median, mean, and SD values for MDA concentrations have been calculated. For statistical analysis of the data, the Wilcoxon two sample test was used to test for statistical differences of MDA plasma concentrations among healthy children, the group of neurological/epileptic children, and the group with cerebellar structural defects.
Results
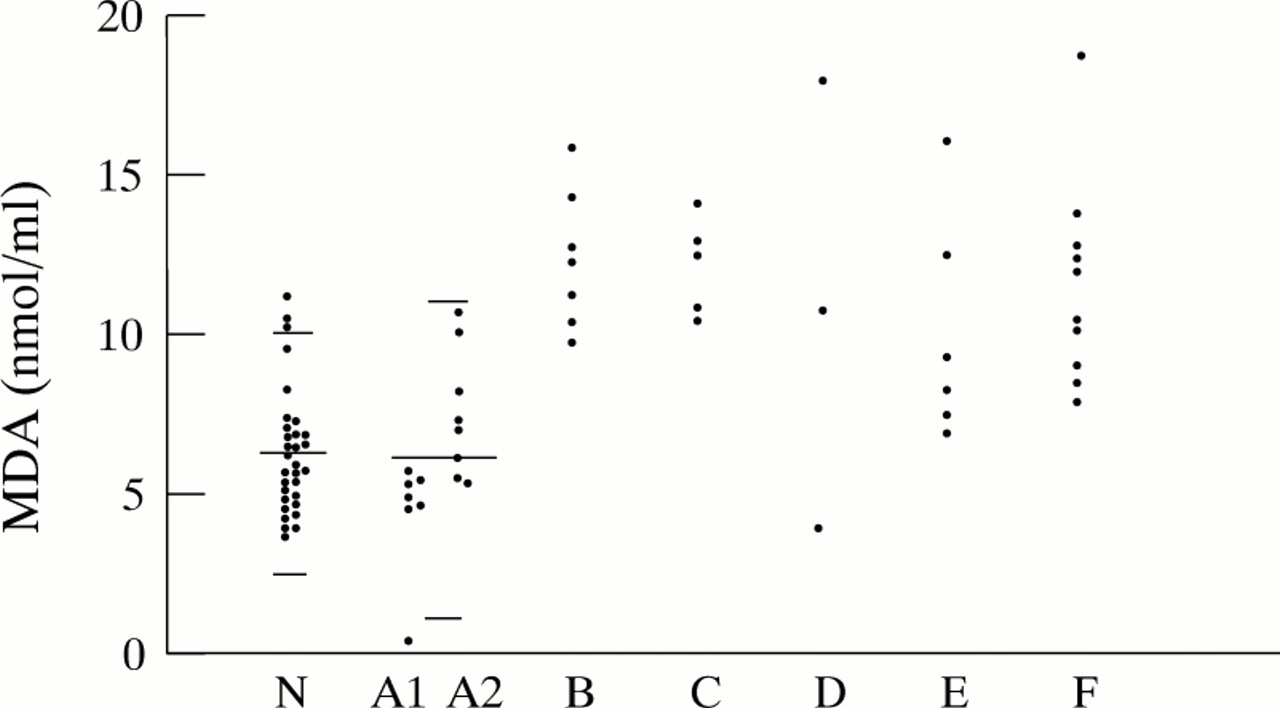
The results of MDA concentrations in plasma with their respective median values have been plotted for each group (fig 1).

The individual values and the median value (horizontal line) calculated for each group have been plotted. Using the Wilcoxon two sample test no significant differences were present among healthy controls and neurological/epileptic disorders (p=0.91). A statistically significant difference was found between the group with cerebellar defects in comparison with healthy controls (p=0.0001), or with the neurological/epileptic group (p=0.0001).
For the control group of 31 healthy children, plasma MDA concentrations showed a mean value of 6.25 nmol/ml with a SD of 1.97 nmol/ml (median value 5.86).
In 15 children with various neurological disorders (epilepsy, extrapyramidal disorders, and undernutrition), in whom neuroimaging had excluded cerebellar involvement, the MDA concentrations with a median value at 5.66 nmol/ml were situated within a similar range as compared with healthy controls (Wilcoxon two sample test non-significant with p = 0.91). Within the group of neurological/epileptic disorders, a subdivision between non-epileptic and epileptic children, corresponding to group A1 and A2 in fig 2, was made. Although all MDA concentrations in the non-epileptic subgroup of seven children (group A1) remained below the calculated mean value for the whole group, in the subgroup A2 of eight children with epilepsy MDA values were found to be increased up to 10.8 nmol/ml.
{kind=link}
{kind=link}
Measured MDA values in nmol/ml in patients with and without pontocerebellar structural abnormalities; group N stands for normal healthy controls, group A1 and A2 represent patients with normal infratentorial structures, where A1 are patients without seizures and group A2 are patients with epilepsy; group B have unilateral cerebellar hypoplasia; group C have midline vermis fusion defects, group D have non-progressive cerebellar hypoplasia; group E have pontocerebellar hypoplasia; and group F represent patients suffering from progressive cerebellar atrophy. The horizontal lines indicate the mean with upper and lower 2 SD values for MDA in the healthy control group and the neurological/epileptic group.
Compared with healthy children and patients with neurological/epileptic syndromes, MDA concentrations for patients with cerebellar structural defects (n=31) were found to be increased significantly (both p values equal to 0.0001; Wilcoxon two sample test). The median value for the cerebellar group was determined at 11.29 nmol/ml. Figure 2 displays the individual MDA concentrations classified according to each subgroup with specific cerebellar structural defects.
MDA values were constantly increased with values ranging between 9.6 and 16.08 nmol/ml in seven tested patients (group B) suffering from moderate or severe mental retardation and unilateral cerebellar hypoplasia of undetermined origin.
MDA values measured in five patients with vermis defects (group C) ranged between 10.55 and 14.25 nmol/ml, that is, in two patients with Joubert’s syndrome, one patient with cerebellar ataxia and cap myopathy, another patient with complete vermis agenesis and mental retardation, and one patient with developmental delay after vacuum extraction.
In the group of three patients with non-progressive and static cerebellar hypoplasia (group D), MDA values were found to be increased in the child with acrocallosal syndrome and the child with the Dandy-Walker variant. In contrast, the MDA value in the child with Dandy-Walker syndrome was normal.
MDA values between 6.96 and 16.28 nmol/ml were found in six children belonging to the group of pontocerebellar hypoplasias or atrophy (group E), that is, pontocerebellar hypoplasia type I and II, PEHO syndrome, and the retarded patient with progressive microcephaly and pontocerebellar atrophy. Only two out of six values were increased well above the upper range of normal, whereas the other four MDA values had an overlap with the normal range in healthy individuals.
MDA measurements in 10 patients belonging to the group with progressive bilateral cerebellar atrophy (group F), showed constantly raised values in eight patients. These comprised two children with infantile and late infantile onset neuronal ceroid lipofuscinosis, one child with Sjögren-Larsson syndrome, one with ataxia telangiectasia, one with Boucher-Neuhäuser syndrome, one with a spastic ataxic syndrome and dysmyelination, and two patients suffering from progressive cerebellar atrophy combined with a peripheral neuropathy. Intermittent increases were found in the two siblings suffering from an autosomal recessive spastic ataxic syndrome with parkinsonian features. For these two siblings, only the increased MDA values have been plotted in figs 1 and 2.
In five out of these 10 patients in group F, there was an overlap of the calculated values as compared with the healthy controls.
Discussion
In an effort to delineate the unclassified or unknown genetic causes of cerebellar syndromes, MDA values in plasma have been used as a screening procedure for detecting increased lipid peroxidation due to oxidative stress.2 3 Oxidative stress is defined as oxygen radical mediated damage to biological material (proteins, lipids, carbohydrates, and DNA) caused by either increased generation and build up of the oxygen radicals (superoxide radical, hydrogen peroxide, and the hydroxyl radical), or due to the diminished removal or inadequate protection against these ubiquitously present oxygen radicals. In aerobic animals, powerful antioxidant enzymes (catalase, glutathione peroxidase, and superoxide oxidases) and radical scavengers (glutathione and vitamin E) are present to remove or scavenge these inorganic and organic oxygen radicals in cell organelles and membranes.2-4 Whenever tissues are damaged, metal complexes will be released from vacuoles, organelles, or other sequestered sites within the cell. Once released, these metal ions can then interact and propagate further lipid peroxidation. Increased MDA can be interpreted as the result of cellular membrane damage initially caused by increased formation of radicals and lipid peroxidation. But increased MDA concentrations can also result from lipid peroxidation, which only accompanies many disease processes where the initial cellular damage and release of metal complexes was elicited by toxins or other causes.1
The cerebellum appears to be more susceptible to oxidative stress than other brain structures. This observation was based upon an extensive search through the literature, from which some defined neurometabolic diseases arose where oxidative stress might be involved in the origin of damage to the cerebellum. Some well known examples associated with cerebellar atrophy are mitochondrial diseases (such as Kearns-Sayre syndrome and MERRF syndrome) or deficiency of respiratory chain enzyme complexes such as cytochrome c oxidase. Other examples are Menkes’ kinky hair disease, spinocerebellar degeneration due to synthetic errors of glutathione (γ-glutamylcysteine synthetase and glutathione synthetase), or conditions with vitamin E depletion due to hypobetalipoproteinaemia or abetalipoproteinaemia. A common denominator in the pathogenesis of cerebellar degeneration in the above mentioned disease entities might be increased oxidative stress.
In mitochondrial disorders the antioxidant enzymes and radical scavengers will fail to overcome oxidative stress because a block in the respiratory chain due to a deficient activity of one of the enzymes involved causes electron leakage and increased superoxide radical production. In patients with complex I deficiency, the subsequent release of free oxygen radical species induces grossly elevated concentrations of mitochondrial manganese superoxide dismutase. However, the antioxidant defences were not capable of preventing free oxygen radical mediated damage as reflected by multiple additional deletions of mitochondrial DNA.10
Likewise, in Menkes’ kinky hair disease the disturbed copper absorption, with resultant lowered activity of the copper dependent enzyme cytochrome c oxidase and cytoplasmic copper zinc superoxide dismutase, causes increased production of the superoxide radical. In conditions with glutathione or vitamin E depletion, the resultant impaired protection against oxygen radicals provokes increased oxidative stress. Part of the clinical correlate for these disorders is a progressive cerebellar degeneration.
It is interesting to note that MDA values were normal in healthy controls and in the majority of neurological patients with normal infratentorial structures, whereas increased MDA was found in Joubert’s syndrome, pontocerebellar hypoplasia types I and II, and in many other patients with progressive cerebellar atrophy of undetermined origin. These preliminary findings suggested the possible contribution of primary or secondary oxygen radical mediated damage to early apoptosis of cerebellar progenitor cells with consequent prenatal or postnatal pontocerebellar maldevelopment or degeneration. We are currently attempting to identify the underlying cause for the observed increase of MDA in some of the patients described in this study.