
Abstract
Summary: A 5-year-old boy with methionine adenosyltransferase (MAT) I/III deficiency, under treatment for the tentative diagnosis of homocystinuria, presented with mildly decreased appetite and sleepiness. MR imaging showed abnormal T1 and T2 prolongations and reduced diffusion in the cerebral white matter. Clinical symptoms and MR imaging findings improved after discontinuation of therapy. We speculate that inappropriate treatment might enhance CNS lesions of MAT I/III deficiency by causing a reversible vacuolating myelinopathy.
Methionine adenosyltransferase (MAT) I/III, expressed solely in the liver, catalyzes the biosynthesis of s-adenosylmethionine from methionine. MAT I/III deficiency, caused by mutations in the MAT1A gene, is characterized by persistent isolated hypermethioninemia (1, 2). Most patients with moderate impairments in MAT I/III activity have no clinical manifestations, but a few with severe impairments present with headache, nystagmus, dysdiadochokinesis, dystonia, and learning difficulties with diffuse T2 prolongation of cerebral white matter (1–3). Those with the highest plasma methionine elevations also have mild to moderate abnormal elevations of plasma total homocysteine, which may lead to a misdiagnosis of homocystinuria due to cystathionine β synthase (CBS) deficiency (4). We report the case of a boy with MAT I/III deficiency treated with CBS deficiency, who presented with mildly decreased appetite, sleepiness, and MR imaging abnormalities in the cerebral white matter. Both the symptoms and the MR imaging findings improved after discontinuation of the therapy.
Case Report
A currently 5-year-old boy was found to have pronounced hypermethioninemia on neonatal screening. In addition to elevated plasma methionine (740 μmol/L; normal, <40), laboratory studies showed elevated total homocysteine (37.4 μmol/L; normal range, 3.0–14.0), leading to a diagnosis of CBS deficiency. At the age of 3 weeks, he began treatments with methionine-restricted diets and oral betaine; pyridoxine was started at age 2.5 years because of difficulty in maintaining an adequately restrictive diet. His early development was normal, and his neurologic examination was unremarkable. During the treatment, plasma methionine was markedly elevated (960–1560 μmol/L), but total homocysteine (6.3–14.9 μmol/L) and other essential amino acids were normal.
After pyridoxine administration, he started to have mildly decreased appetite and sleepiness in the daytime. Neurologic examination was unremarkable, with the patient’s IQ measuring 121 at 3.5 years old. The results of routine laboratory studies on blood and urine were either within normal limits or negative. MR imaging was performed at 3 years, 10 months to evaluate for possible cerebral lesions. Spin-echo T1-weighted (440/9 [TR/TE], Fig 1A) and fast spin-echo T2-weighted images (Fig 1B) revealed diffuse T1 and T2 prolongation in subcortical areas extending to deep white matter with relative sparing of the corticospinal tracts, corpus callosum, optic radiations, ventral brain stem, and cerebellar white matter. Symmetrical lesions in the dorsal brain stem were also recognized from the red nucleus to olivary nucleus, which suggested involvement of the central tegmental tracts (Fig 1D). Diffusion-weighted imaging (spin-echo echo planar imaging, TR: 5000; b = 1000) showed high signal intensity, and an apparent diffusion coefficient (ADC) map demonstrated decreased ADC value of the lesion (Fig 1C, ADC = 0.553 × 10−3 mm2/s in the frontal white matter [arrowhead], as compared with an age-matched normal value of 0.860 ± 0.040 × 10−3 mm2/s). These findings were suggestive of MAT I/III deficiency rather than CBS deficiency. Analysis of MAT 1A gene revealed a compound heterozygote for two MAT1A mutations: 274T→C (Y92H) from his father and 1067G→C (R356P) from his mother, which led to a diagnosis of MAT I/III deficiency. After discontinuation of the therapy at 4 years of age, his clinical symptoms disappeared. Plasma concentration of methionine and total homocysteine were 570–1090 μmol/L and 14.0–22.0 μmol/L, respectively. Follow-up MR imaging at 5 years demonstrated normalization of white matter signal intensity (Fig 1E and F). The ADC value also recovered to subnormal range (ADC = 0.775 × 10−3 mm2/s). Symmetric T1 and T2 prolongation was still observed in the central tegmental tract.
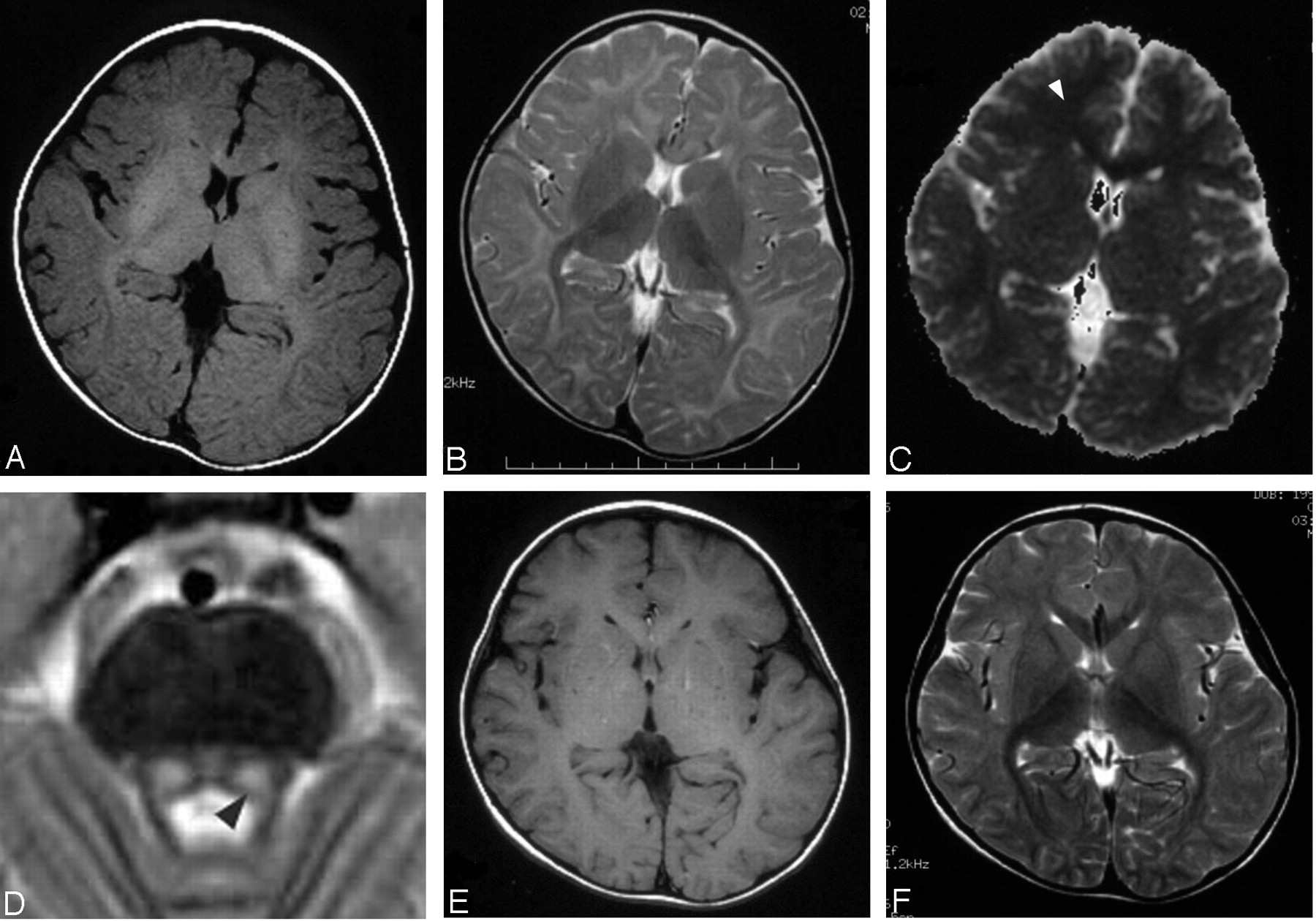
MR imaging at 3 years showing T1 and T2 prolongation (A, B) and reduced diffusion (C; ADC = 0.553 × 10−3 mm2/s in the frontal white matter [arrowhead]) in the white matter, and T2 prolongation in the symmetric central tegmental tract (D, arrowhead). Patchy foci of T1 and T2 shortening (E, F) were observed in the white matter at 5 years.
Discussion
In MAT I/III deficiency, diagnostic uncertainty or error may arise when the abnormal elevations of plasma total homocysteine are combined with elevated methionine; this combination of abnormalities often leads to a diagnosis of CBS deficiency and to prolonged use of methionine-restricted diets and other interventions as in the present and other reported patients (4). A causal relationship between MAT I/III deficiency and abnormalities of cerebral white matter at MR imaging has been suggested by the beneficial effect of s-adenosylmethionine for one patient (3) and by the fact that patients with other metabolic disorders that lead to subnormal s-adenosylmethionine in the brain are prone to white matter abnormalities (3). The available evidence suggests that pathophysiological consequences are more likely to result from s-adenosylmethionine deprivation than from elevated methionine (4).
Improvement of MR imaging findings and clinical symptoms after discontinuation of therapy in this patient suggested that these abnormalities might be associated with the therapy. The concentration of methionine during therapy was higher than that after its discontinuation; this probably represents a response to betaine therapy. Although concentration of s-adenosylmethionine was not measured, elevation of methionine would be expected to increase the rate of the methionine conversion to s-adenosylmethionine catalyzed by any residual activity of MAT I/III. Clinical and radiologic improvement, however, would not be explained by an increase of s-adenosylmethionine concentration related to treatment. A previously reported neonate with MAT I/III deficiency became apneic and unresponsive and required respiratory support after administration of pyridoxine but improved dramatically after discontinuation of pyridoxine (4). We accordingly speculate that, in both that case and the case reported here, pyridoxine treatment might have worsened the abnormalities caused by the MAT I/III deficiency. Another possible explanation for clinical worsening during treatment is a competition between methionine and other neutral amino acids for transport into brain, combined with elevated methionine in response to betaine therapy and falling levels of other essential amino acids under restrictive diet, leading to a critical deficiency of neutral amino acids in brain. In any case, the precise diagnosis should be pursued to avoid the possible adverse consequence of inappropriate treatment.
Complete resolution of the white matter abnormalities after treatment with s-adenosylmethionine was reported in one patient with MAT I/III deficiency (3). Therefore, the white matter lesions of MAT I/III deficiency may be potentially reversible. The reduced diffusion of the white matter that we noted might give a clue to the pathogenesis of the lesion. Reduced diffusion can be observed in cytotoxic edema due to cellular energy failure, such as acute infarction or encephalopathy, but this was clinically unlikely for this patient. Recently, decreased ADC values of white matter lesions have been observed in patients with Canavan disease, nonketotic hyperglycinemia, and phenylketonuria (5–7). A possible explanation for the reduced diffusion in these disorders is intramyelinic edema as a result of separation of myelin layers, so-called vacuolating myelinopathy (5–7). Results obtained with experimental animals suggest a connection between deficient MAT activity and myelin abnormalities. Administration of cycloleucine, an inhibitor of MAT, causes decreases of proteolipid protein and dimmer-20, major components of myelin in CNS of rats (8). Light and electron microscopic examination after administration of cycloleucine reveals that intramyelinic vacuolation develops in the white matter of mice (9). The morphologic effects are considered to be the result of s-adenosylmethionine deficiency impairing a transmethylation of myelin basic protein, a process known to be important in the formation and stabilization of myelin (9). Despite an absence of pathologic data in patients with MAT I/III deficiency, it is reasonable to speculate that the reduced diffusion of the white matter may also reflect vacuolating myelinopathy, although the mechanism by which pyridoxine may worsen the myelin abnormality is uncertain.
Another MR finding in this patient was symmetrical involvement of the central tegmental tract. Lesions of the central tegmental tract have been noted in severely asphyxiated infants, in patients with leukoencephalopathy with vanishing white matter, and in nonketotic hyperglycinemia (7, 10), but have never been mentioned in MAT I/III deficiency. Clinically, involvement of the central tegmental tract results in palatal myoclonus and an uncontrollable tremor, presumably caused by loss of inhibitory control after focal lesions to the dentato-rubro-olivary pathway. Tremor was observed in a previously reported patient (1), but the present patient had no such symptoms. Further study is needed to clarify the frequency of involvement of central tegmental tract and its clinical significance in patients with MAT I/III deficiency.
Acknowledgments
We thank Dr. M. Nagao, of Department of Pediatrics, National Nishi-Sapporo Hospital, Sapporo, Japan, for analyzing the MAT1A gene. We appreciate the excellent technical support provided by Y. Nakano, F. Morita, and T. Isobe.
References
- Received December 30, 2003.
- Accepted after revision March 5, 2004.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.