Article Text

Abstract
Although neurological symptoms are common in Whipple's disease, patients rarely have a purely neurological presentation and involvement restricted to the central nervous system is uncommon. A 39 year old woman presented with a meningoencephalitic illness, which responded to penicillin. Eleven months later she developed recurrent stroke-like episodes. Patchy enhancing meningeal, cortical, and subcortical lesions thought to be vascular in origin developed within nine days of the onset of symptoms. No evidence was found of a cardiovascular source of emboli, vasculitis, or thrombophilic condition. A brain biopsy showed meningoencephalitic features suspicious of Whipple's disease associated with leptomeningeal arterial fibrosis and thrombosis. DNA polymerase chain reaction confirmed Tropheryma whippelii in both blood and brain tissue. The neurological manifestations of cerebral Whipple's disease are varied and very rarely include stroke-like symptoms. The pathogenesis of cerebral infarction in Whipple's disease is not well established but arterial fibrosis and endocarditis complicated by embolisation have been reported. This case emphasises the importance of early brain biopsy in unusual cases of stroke and illustrates the clinical utility of polymerase chain reaction to confirm Whipple's disease.
- Whipple's disease
- Tropheryma whippelii
- cerebrovascular disease
- stroke
- polymerase chain reaction
- ACE, angiotensin converting enzyme
- CSF, cerebrospinal fluid
- CT, computed tomography
- IQ, intelligence quotient
- MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like syndrome
- MERRF, myoclonic epilepsy with ragged red fibres
- MR, magnetic resonance
- OMM, oculomasticatory myorhythmia
- OFSM, oculofacial skeletal myorhythmia
- PCR, polymerase chain reaction
- WD, Whipple's disease
Statistics from Altmetric.com
- ACE, angiotensin converting enzyme
- CSF, cerebrospinal fluid
- CT, computed tomography
- IQ, intelligence quotient
- MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like syndrome
- MERRF, myoclonic epilepsy with ragged red fibres
- MR, magnetic resonance
- OMM, oculomasticatory myorhythmia
- OFSM, oculofacial skeletal myorhythmia
- PCR, polymerase chain reaction
- WD, Whipple's disease
Whipple's disease (WD) is a chronic inflammatory multisystemic disorder with prominent but not invariable gastrointestinal involvement. It is caused by the Gram positive bacillus Tropheryma whippelii, large numbers of which accumulate within macrophages in the mucosa of the small bowel and in other affected tissues.1 The classic clinical features include chronic diarrhoea with malabsorption, abdominal pain, relapsing-remitting migratory polyarthralgia, lymphadenopathy, weight loss, hyperpigmentation of the skin, and fever of unknown origin.1–3 A variety of neurological signs and symptoms, including on very rare occasions a stroke-like presentation, have also been described in a significant minority of infected patients, usually in concert with other systemic symptoms.4,5 The conclusive diagnosis of WD has long been hampered by an inability to culture the organism, which required reliance on the ultrastructural recognition of rod shaped bacilli with a characteristic trilaminar membrane.1,6 The more recent use of polymerase chain reaction (PCR) based assays to identify and characterise the organism has significantly improved the diagnosis and management of WD.1,6 We report on a patient with a stroke-like presentation where PCR facilitated the diagnosis of cerebral WD and review the cerebrovascular aspects of this disease.
CASE REPORT
A 39 year old left handed woman was well until January 1999, when she developed a petechial rash with meningism associated with reduced consciousness and diarrhoea. Her clinical state deteriorated as she developed a left basal pneumonia with evidence of disseminated intravascular coagulation. A lumbar puncture was omitted as a computed tomographic (CT) brain scan suggested diffuse cerebral oedema. No causal organism was isolated and the purpuric rash was not sampled. Treatment was largely supportive but empirical high dose intravenous benzylpenicillin was given for presumed meningococcal septicaemia. She made an excellent functional recovery but felt irritable and depressed. Her father and sister had amyotrophic lateral sclerosis.
On 19 December 1999, while on holiday abroad, she experienced weakness of the right arm and face with slurring of speech lasting a few seconds with complete recovery. She then felt generally unwell and two days later developed weakness of the right arm, which lasted for an hour. That same evening both legs went weak for a few minutes but she was able to walk to a local general practitioner. After returning to her hotel she collapsed and transiently lost consciousness. On 22 December she had a fleeting episode of left arm paraesthesia with urinary incontinence and an hour later she developed a moderate right hemiplegia and memory impairment. On 24 December she was found to have a left hemiplegia, which was followed by a generalised seizure. She subsequently had difficulty speaking and started to receive phenytoin. Repeat CT head scans during this period were normal. An intermittent mild pyrexia was noted. On 28 December she was started on intravenous heparin for presumed cortical venous thrombosis and was transferred by air ambulance to our unit.
On arrival she was apyrexial and drowsy but could follow simple commands. There was no spontaneous speech and she had a mixed dysphasia. Her voluntary eye movements were difficult to assess but appeared normal. The oculocephalic and other brainstem reflexes were intact. She had a severe spastic quadriparesis without spontaneous limb movement. There was no clinical evidence of endocarditis, arthritis, or orogenital ulceration.
Full blood count and clotting screen were normal. The serum albumin was low at 32 g/l (normal 36–50 g/l) and aspartate transaminase slightly high at 44 U/l (normal 13–42 U/l). The erythrocyte sedimentation rate was increased at 36 mm/h and the C reactive protein was high at 48 mg/l (normal < 5 mg/l). Serum angiotensin converting enzyme (ACE) concentration was normal. Antineutrophil cytoplasmic antibody, lupus anticoagulant, anticardiolipin antibody, protein S, protein C, antithrombin III, and autoantibodies were all normal or negative. A lumbar puncture had a normal opening pressure and the cerebrospinal fluid (CSF) lactate concentration was 3.4 mmol/l (normal 0.5–2.2 mmol/l) with a raised CSF ACE concentration of 17.4 U/l (normal < 2 U/l). Routine CSF tests (including cytology and tuberculosis culture) were normal and there were no oligoclonal bands. Genetic testing for the common mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like syndrome (MELAS) and myoclonic epilepsy with ragged red fibres (MERRF) was negative. Serological tests for HIV, Borrelia spp, and syphilis (Treponema pallidum haemmaglutination assay) were all negative.
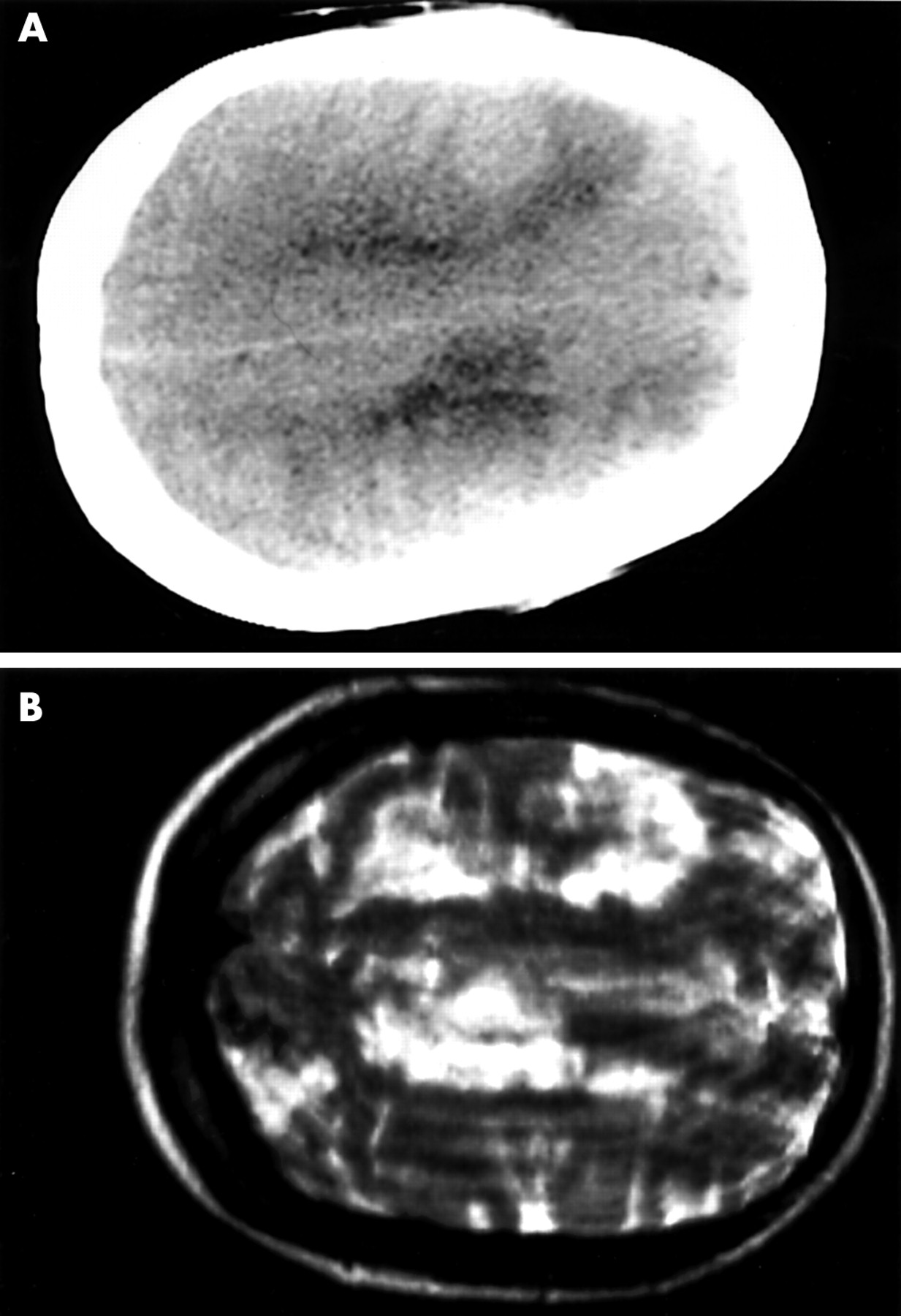
The previous CT head scans were confirmed as normal. A further CT head scan showed patchy bilateral low density areas in the upper frontal and parietal lobes in the grey and white matter (fig 1A). Magnetic resonance (MR) imaging (29 December) showed bilateral hemispheric high signal changes on T2 weighting (fig 1B). MR angiogram and venogram were normal. A repeat MR study on 12 January 2000 showed gadolinium enhancing meningeal and parenchymal high signal areas in both hemispheres with ventricular dilatation. An electroencephalogram showed a pronounced excess of slow (δ) waves over both hemispheres with no epileptic activity. Conventional catheter cerebral angiography was normal and a transoesophageal echocardiogram showed no cardiac abnormality.

(A) Axial computed tomography of the head (28 December 1999) with patchy low density changes in both frontoparietal lobes. (B) Axial T2 weighted magnetic resonance image (29 December 1999) showing high signal lesions in both hemispheres.
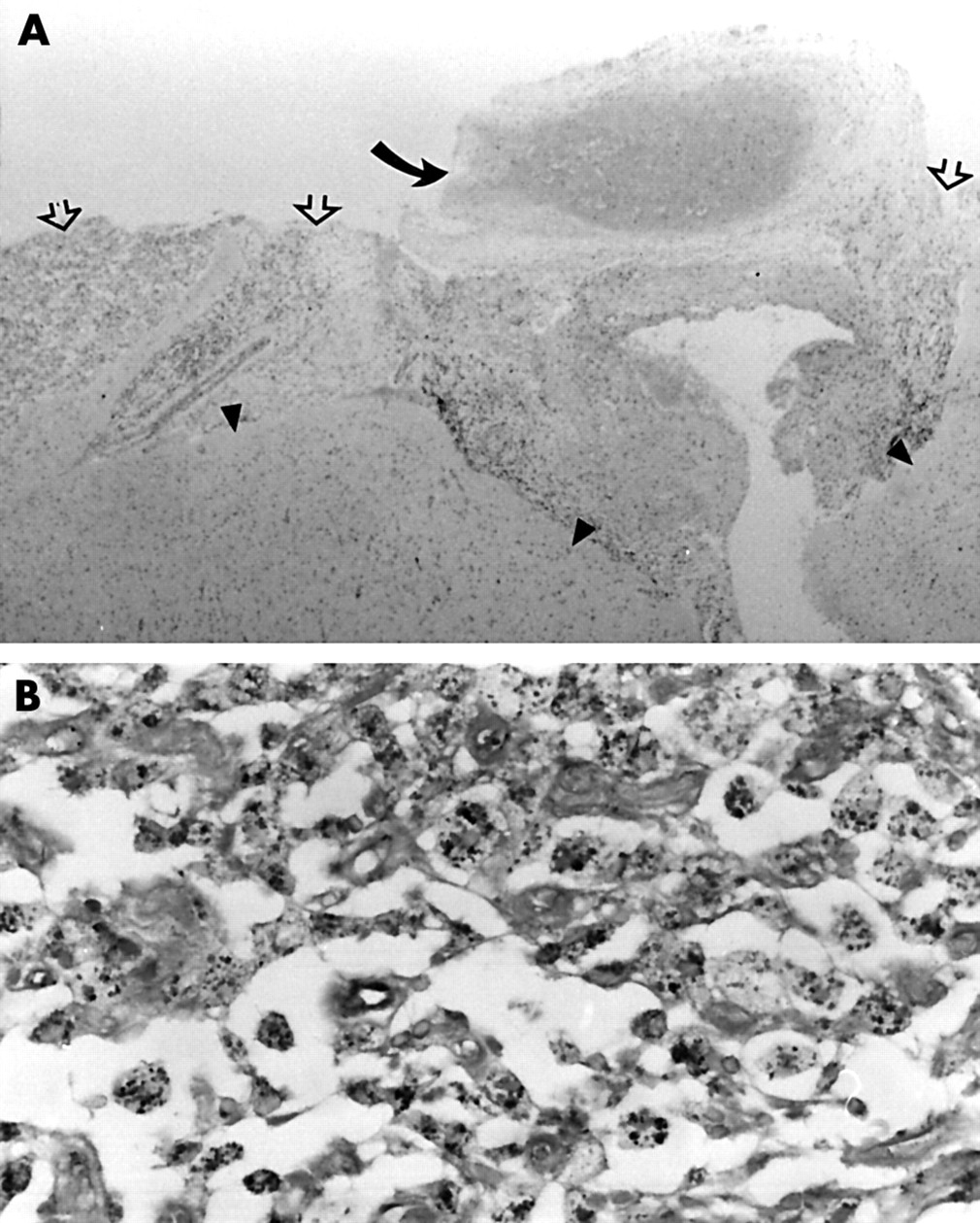
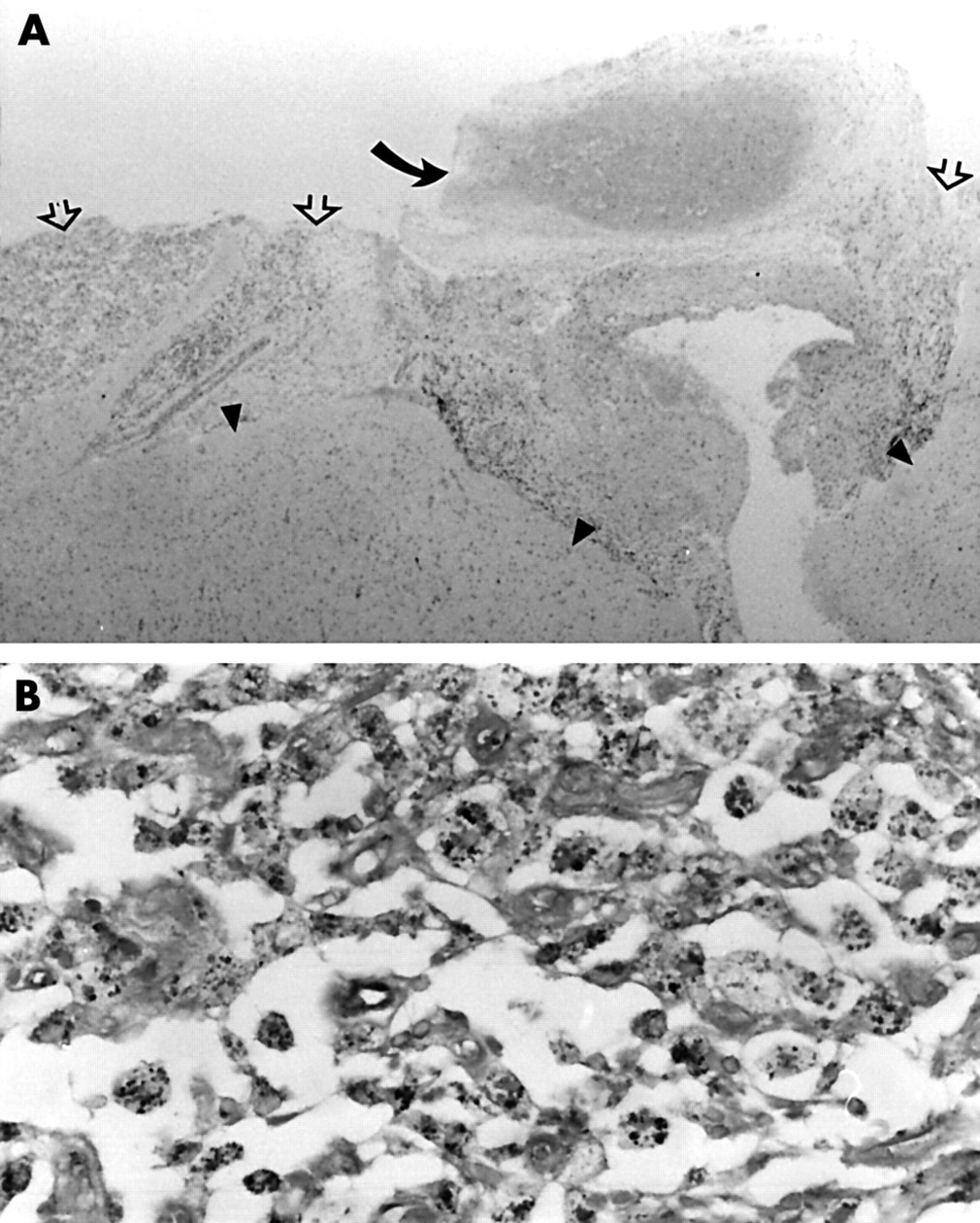
She underwent brain biopsy on 24 January 2000 after no clinical improvement. This showed meningoencephalitic features with a predominantly leptomeningeal infiltrate of foamy macrophages and leptomeningeal arterial fibrosis and thrombosis without obvious active vasculitis (fig 2A). Gram and Ziehl-Neelson staining did not show bacterial or fungal organisms but periodic acid Schiff and silver (Grocott) stains highlighted granular material and occasional curvilinear forms within the macrophages (fig 2B). Electron microscopy showed phagocytic vacuoles and secondary lysosomes containing amorphous debris and membranous profiles. No intact bacillary forms were identifiable. These findings were considered non-diagnostic but suggestive of WD. The diagnosis was confirmed by PCR when both blood and brain specimens were positive for T whippelii DNA encoding the 16S ribosomal RNA. PCR was performed at a national reference centre using two sets of primers (W3FE,W2RB 59% sensitivity and W3AF,W4AR 96% sensitivity). A CSF specimen was PCR negative. An ultrastructurally normal jejunal biopsy was not subjected to PCR analysis.
{kind=link}
{kind=link}
Microscopy of brain biopsy. (A) Leptomeningeal inflammatory infiltrate (open arrows) and organising thrombosis in a fibrosed artery (curved arrow). Underlying cortex (arrow heads) are gliotic with increased numbers of microglial cells (haematoxylin and eosin, original magnification ×25). (B) Detail of leptomeningeal inflammatory infiltrate. Foamy macrophages filled with Grocott positive particulate material (Grocott, original magnification ×400).
An indefinite course of high dose oral cotrimoxazole was commenced and further MR scanning on 28 February showed less enhancement with slight resolution of the previous high signal areas on T2 weighting. Speech has recovered and she became fluent with no dysphasic errors. Memory improved greatly and formal neuropsychological testing gave a verbal intelligence quotient (IQ) of 63 (predicted premorbid full scale IQ 87). Mood and speech were occasionally inappropriate and a supranuclear gaze palsy was noted. Unfortunately there had been very little motor recovery and she has been left with a severe spastic quadriparesis but she can feed herself with help. She is able to use an electric wheelchair.
DISCUSSION
The central nervous system (CNS) may be affected in as many as 43% of patients with WD during the course of their disease.7–9 A neurological presentation is less common (5%) and is often followed by disease confined to the CNS.3,5,10 Subclinical gastrointestinal disease may, however, have been underrecognised in some of these cases because of suboptimal biopsy procedures.4 CNS involvement carries the highest risk for relapse.11 Cognitive changes (71%), supranuclear gaze palsy, and altered consciousness are the commonest neurological findings.4 Other CNS signs include oculomasticatory (OMM) and oculofacial skeletal myorhythmia (OFSM), myoclonus, ataxia, hypothalamic dysfunction, cranial nerve abnormalities, and sensory deficits.4,5 Although upper motor neuron signs have been found in as many as 37% of patients with CNS disease, clinical reports of stroke-like symptomatology are extremely rare.4,12
The neuropathology of cerebral WD is that of a disseminated or focal macrophagic encephalitis or meningoencephalitis favouring subpial and subependymal grey matter.13,14 Mass lesions and obstructive hydrocephalus may occasionally complicate the pathology. Infarcts are also described but their frequency is unclear and associated features are not always detailed or referenced. Leptomeningeal involvement may occasionally be associated with small infarcts, usually < 5 mm in diameter, scattered throughout the brain.13 Arterial or arteriolar fibrosis and thickening have been suggested as a cause of infarction in affected parts of the brain.14 Whether the latter is secondary to surrounding chronic inflammation or to a primary vasculitic process is uncertain. Vasculitis has been described in retinal WD as a primary event as well as part of a Jarisch-Herxheimer reaction following antibiotic treatment but has not been reported elsewhere in the CNS.15,16 A single case of recurrent stroke due to embolisation from T whippelii endocarditis has been described.12 Thirty per cent of patients have murmurs and postmortem studies suggest an even higher incidence of endocarditis.17–19 A cardiac source could not be identified in our case and histology does not permit distinction between in situ thrombosis and thromboembolisation.
Louis et al4 have proposed guidelines for the diagnosis of CNS WD based on the findings of their extensive review of all previously reported cases. A definite diagnosis requires the presence of OMM or OFSM or a positive biopsy or positive PCR analysis. Neurological signs are required when the positive results have been obtained from non-CNS tissue. Given the infrequent incidence of OMM and OFSM (and some doubt about its specificity), diagnosis is mostly dependent on laboratory investigations.
The majority of intestinal (70%), brain (83%), and all (also including lymph node and vitreous fluid) biopsies (89%) performed in the cases reviewed by Louis et al were regarded as diagnostic.4 The limitations posed by focal CNS disease and inaccessible sites may be successfully addressed by stereotactic biopsy procedures.20
Electron microscopy is being supplanted by PCR analysis as the confirmatory diagnostic test complementary to biopsy due to its greater potential sensitivity and ease of application.1,6,21 Some concerns about the disease specificity of positive PCR results, however, raise caution about reliance on it alone to make a definitive diagnosis.22 The presence of T whippelii DNA in the saliva of healthy volunteers (35% in one series) and in duodenal biopsy (4.8%) and gastric juice (11.4%) gastroscopy specimens from patients with no clinical signs of WD suggests that the organism may be a normal commensal agent.22,23 The use of two sets of well characterised primers and the analysis of preferably more than one tissue substrate have been advised to maximise sensitivity and specificity.1 Our patient's biopsy showed changes suggestive of WD, PCR was positive in both brain and blood specimens using two sets of primers, and a definite improvement followed treatment with cotrimoxazole. PCR may also be useful to monitor response to treatment and prognosis.1,21,24,25
Eighty per cent of patients with neurological symptoms and 70% of patients without neurological symptoms have yielded positive CSF PCR results in one series.24 The observation that CSF PCR may be more sensitive in the presence of CSF pleocytosis may explain the negative PCR result in our case.26 The significantly raised CSF ACE concentration may be accounted for by macrophage activation. Increased serum ACE concentrations have been described in isolated cases of WD.27
This case emphasises the importance of early cerebral biopsy and the need to consider WD in cases with atypical stroke-like episodes. In retrospect a brain biopsy should have been done earlier once the negative blood test, angiography, and echocardiogram had been obtained. The application of PCR techniques specific to and sensitive for T whippelii promises greater recognition of CNS involvement and may enhance our understanding of WD's pathogenesis.
Acknowledgments
We thank Drs N Clitherow, T Nixon, and T Smith of the Neuroradiology Department at the Walton Centre for interpreting all images. We are also grateful to Drs N P Mapstone and S A Misbah, Yorkshire Regional Immunology Service and Molecular Pathology Laboratory, Leeds General Infirmary, who performed the Tropheryma whippelii PCR analysis.
This case was presented at the World Congress of Neurology XVII in June 2001.
REFERENCES
Footnotes
-
Competing interests: none declared