Article Text

Abstract
The main clinical features of CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy) are stroke, dementia, and migraine. A reversible acute encephalopathy was the principal presentation in six of 70 patients in a British prevalence study. The episodes lasted seven to 14 days, presenting with fever, acute confusion, coma, and fits; there was full recovery but in two cases identical episodes recurred some years later. All patients had a previous history of migraine with aura and were originally misdiagnosed as viral encephalitis. CADASIL should be considered in acute unexplained encephalopathies. MRI white matter changes, previous migraine with aura, and a family history of stroke and dementia may be useful pointers to the diagnosis.
- encephalopathy
- CADASIL syndrome
- dementia
Statistics from Altmetric.com
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) causes lacunar strokes, migraine, and dementia. The disease is caused by mutations in the Notch3 gene on chromosome 19, with a prevalence of at least 1 per 100 000 (Markus H, British CADASIL study, unpublished data). There have been two large clinical studies with 102 and 105 patients.1,2 The first symptom in the review paper by Desmond et al was stroke or transient ischaemic attacks in 43% of patients, migraine in 40%, depression in 9%, and cognitive impairment in 6%.3 Epilepsy has been reported in 2–10% of patients, most often following strokes.
We describe an acute encephalopathic presentation of the disease in six patients (10% of our series). All cases were identified as part of a British CADASIL prevalence study and had Notch3 mutations.
CASE 1
A 37 year old man developed migraine without aura as a child. When he was 29 his migraines changed in character, with a prolonged aura and right sided hemisensory symptoms lasting one to two hours, and hemianopia lasting up to several hours. His mother had migraine and died aged 53 with dementia.
At age 34 the patient developed fever and headache, and the following day he became confused and hallucinated. He became drowsy with left sided weakness and a left homonymous hemianopia which progressed over 48 hours. Magnetic resonance imaging (MRI) of the brain showed multiple peripheral and periventricular white matter lesions. Cerebrospinal fluid (CSF) was normal apart from a raised protein of 0.8 g/l. On the fifth day he developed frequent aversive seizures to the left, followed by expressive dysphasia. He was treated with intravenous phenytoin. An electroencephalogram (EEG) showed slow wave activity over the left hemisphere and an almost flat record on the right. He made a slow recovery over two weeks and left hospital with minimal dysphasia. Neurological examination two years later was entirely normal. Genotyping demonstrated the C583T mutation.
CASE 2
This was a 41 year old woman who developed migraine with sensory and visual auras when aged 14. Her mother also had migraine with aura from her 20s, and a stroke aged 67.
When she was 34 the patient developed the first of her two attacks of encephalopathy which started with a typical migraine attack, but the headache, flashing lights, and right sided tingling persisted and were followed by confusion. She was admitted to hospital after four days, where she was drowsy with a mental test score of 7/10, mild right sided weakness, and bilateral extensor plantar responses. CSF examination was normal. An EEG showed bilateral delta waves. MRI of the brain was abnormal with extensive patchy high signal change throughout the deep white matter of both cerebral hemispheres. A diagnosis of encephalitis was made and she was treated with dexamethasone, acyclovir, and broad spectrum antibiotics. She had one tonic–clonic seizure followed by a Todd’s paresis on the right the day after admission. This was treated with intravenous phenytoin and no further seizures occurred. She slowly recovered over 10 days and returned to her clerical job a few weeks later with no apparent deficits.
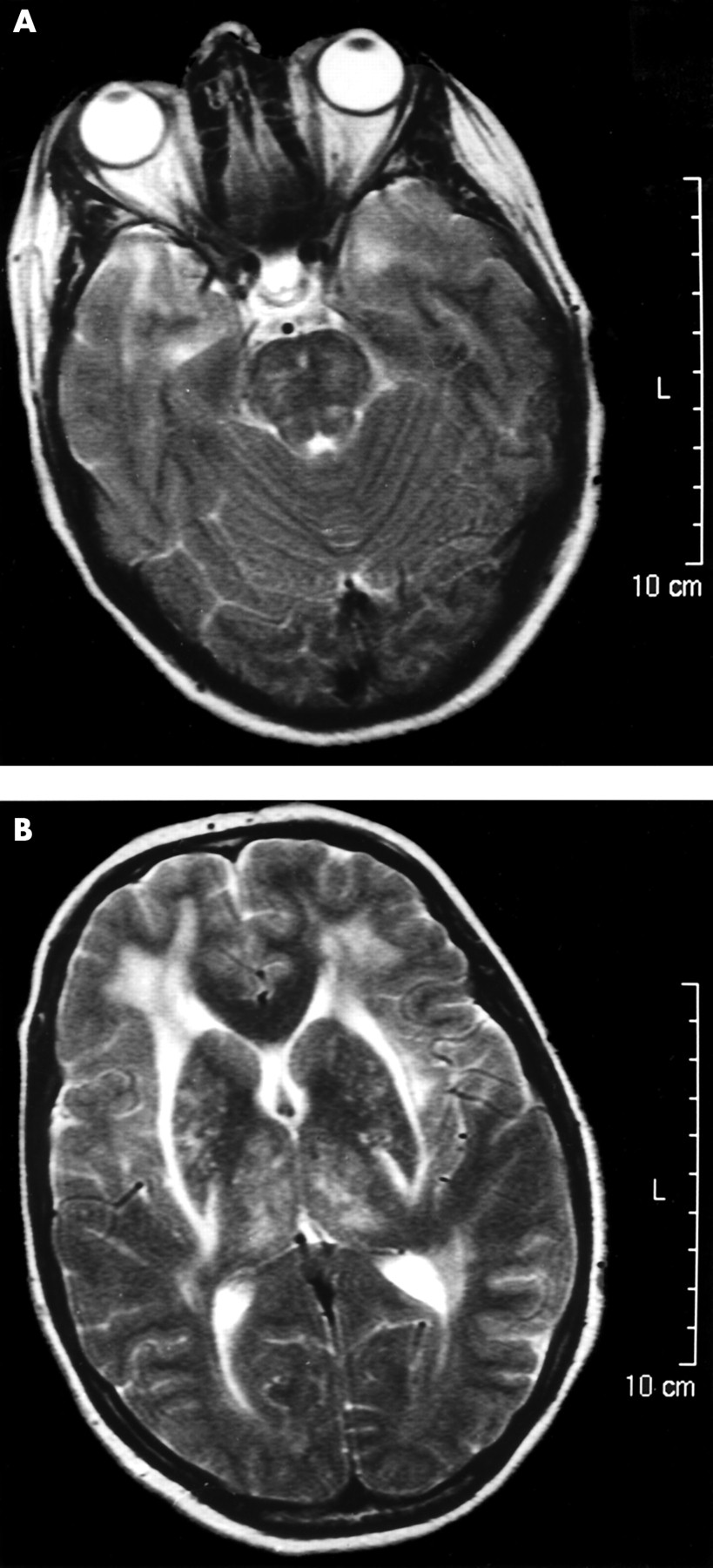
She remained well for seven years until she was readmitted with her second episode when aged 41, with a two day history of severe migraine headache, right arm weakness, vomiting, and blurring of vision. She was mildly pyrexial (37.5°C), with a Glasgow coma score of 13/15, a mild right hemiparesis and right homonymous hemianopia, brisk deep tendon reflexes, and bilateral extensor plantar responses. She was treated with antibiotics and acyclovir. Repeat MRI scan again showed extensive white matter disease, this time involving the centrum semiovale and the anterior poles of the temporal lobes (fig 1⇓). This led to a diagnosis of possible CADASIL. She again suffered two generalised tonic–clonic seizures and was treated with phenytoin. She made a gradual recovery over nine days and returned to work. She has remained asymptomatic during 12 months of follow up. Genotyping demonstrated the C622T mutation.

{kind=link}
MRI images from case 2 showing features characteristic of CADASIL. (A) Image through the temporal lobes showing confluent high signal in both temporal poles. (B) Image through the basal ganglia showing confluent high signal in the external capsules.
CASE 3
This 53 year old man had a 20 year history of migraine with sensory aura. His father had migraines and developed dementia in his 50s. In April 1999 the patient developed right sided numbness akin to his migraine aura but then lost coordination and developed a headache. The next day he was almost unrousable and was admitted to hospital, where he was noted to have headache, fever, confusion, a left homonymous hemianopia, and left sided inattention. CSF examination and computed tomography (CT) of the brain were normal. An EEG showed marked slowing over the right hemisphere. On MRI there was extensive high signal affecting the deep and subcortical white matter of both cerebral hemispheres. He was diagnosed as having viral encephalitis and treated with acyclovir. He became increasingly comatose over five days but then slowly made a full recovery and was discharged after 13 days. His memory and attention were slightly impaired after the acute illness but in retrospect he had probably had mild cognitive symptoms before the acute episode. Genotyping demonstrated the CT293JT mutation.
CASE 4
This 62 year old woman had a history of migraines with sensory aura from her 20s. Her mother had migraines and severe depression, and died with progressive dementia in her 60s.
At age 59 the patient developed a headache, became confused and disorientated, and was admitted with increasing coma and fever but no localising neurological findings. CT of the brain and CSF examination were normal, an EEG showed right temporal slow wave activity, and a MRI showed symmetrical bilateral white matter high signal. She was diagnosed as a case of possible herpes simplex encephalitis and treated with acyclovir. Eleven days later she made a full recovery except that she has had four migraines followed by periods of confusion and disorientation lasting one to three days, with full recovery. Genotyping demonstrated the C622T mutation.
CASE 5
This 61 year old woman developed migraines with visual or sensory auras when aged 23. Her father had the first of a series of strokes age 48 and died aged 60. The patient presented at 38 years of age with the first of three episodes of encephalopathy which started with two days of disorientation and numbness in the left foot. She was unable to name family members and her speech became sparse and of limited content. She was admitted to hospital where she developed visual hallucinations involving the disappearance of the lower halves of people’s bodies and confusion, but she made a full recovery after two weeks.
At age 50 she had a second almost identical episode, again being admitted to hospital for two weeks with visual hallucinations and confusion.
When aged 60 she was admitted with a third episode preceded by diarrhoea and vomiting and a throbbing headache, further vomiting, and again the development of confusion and visual hallucinations of lower body disappearance. During the 10 day admission, she had eight aversive seizures with head turning to her right and loss of awareness, followed by postictal right hand weakness. She was treated with intravenous phenytoin and made a complete recovery.
When examined at age 61 the only findings were minimally increased tone in the legs and a left extensor plantar response. The mini-mental test score was normal. MRI of the brain showed confluent high signal white matter lesions on T2 weighted imaging. Genotyping demonstrated the C583T mutation.
CASE 6
This 36 year old woman had a history of migraine with aura involving visual scintillations for 15 minutes, followed by speech problems for 15–30 minutes and a mild throbbing headache. Her mother was case 5, and there was a family history of premature stroke. Age 30, when 38 weeks pregnant with her second child, this patient had a migraine aura without headache involving right sided numbness and speech problems lasting two hours. Because a transient ischaemic attack was considered she underwent urgent caesarean section. Five days later she became drowsy and confused, and although rousable was only able to obey simple commands. After another two days she suffered a single major seizure. She slowly recovered and was discharged after 10 days. Her initial diagnosis was viral encephalitis and she was treated with acyclovir. CT of the brain showed only basal ganglia calcification. CSF examination was normal and her EEG showed left anterior spike discharges with right hemisphere slow waves. Anticardiolipin antibody was found to be positive, and the alternative diagnosis of intracranial venous thrombosis was considered. When seen, following the diagnosis of CADASIL in her mother, neurological examination and mini-mental testing was normal. She had the same Notch3 mutation as her mother (C583T).
DISCUSSION
These six cases all suffered an acute encephalopathic illness rather than any of the commonly described presentations of CADASIL. In all, a misdiagnosis of acute encephalitis was made and the correct diagnosis of CADASIL delayed, in some cases for many years.
The cardinal features of these cases were confusion in all six, headache at onset in four, pyrexia in four, fits in four, full recovery over seven to 14 days in all cases, and recurrence of virtually identical attacks in two patients. Fits, which were seen in four of the six cases, were both generalised and focal. The fits always developed some days after the onset of coma, so could not easily be the cause of the confusional state. All cases had a history of migraine with aura, and the encephalopathic episodes seemed to start with an otherwise typical migraine attack.
A useful pointer to the diagnosis was the presence of white matter abnormalities on MRI in all our six patients. Recent studies have shown that involvement of the anterior temporal pole is a sensitive and relatively specific marker for CADASIL, while involvement of the external capsule and corpus callosum are common.4,5 In four cases there were marked anterior temporal changes and in six the external capsule was involved.
The association of confusion, hallucinations, and coma with migraine is best described in familial hemiplegic migraine (FHM). In 1985 Fitzsimons and Wolfenden6 wrote about a family with migraine associated with prolonged coma and cerebral oedema which could follow trivial head injuries and could be fatal. This family also had an associated autosomal dominant cerebellar ataxia. Recently Kors et al described three patients with FHM associated with a novel mutation in the CACNA1A calcium channel subunit gene, in which delayed cerebral oedema and fatal coma occurred after minor head injury.7 There is now increasing evidence linking FHM, episodic ataxia type 2, and SCA6 to mutations in the same gene, the exact phenotype of which may at least in part be determined by the differing mutations.8,9 Only half the cases of FHM are linked to mutations in the CACNA1A gene on chromosome 19, and these families have some degree of cerebellar disease. Linkage to chromosome 1 has been described by Echenne et al in a case of FHM without cerebellar features.10
The mechanism underlying CADASIL associated acute encephalopathy is unknown but the remarkable similarities to some features of the calcium channel gene mutations make some indirect channel disorder seem likely, and raises the possibility that acetazolamide might be a possible treatment.
Such episodes of acute resolving encephalopathy associated with CADASIL have only rarely been described. Chabriat et al reported an unusual family in which four of 12 family members had probably suffered similar attacks.11 Their case 1.1 died after an eight day episode of confusion and coma, case 2.1 had two episodes of stupor, case 2.3 suffered 24 hours of confusion with a slow EEG, and case 2.6 had a sudden headache followed by coma and fevers and was admitted to intensive care, had 500 white cells in her CSF, and made a full recovery after 15 days. This family is now known to have an unusual splice site mutation.12 However, all of our six cases had the more common point mutations.
In 2002, Feuerhake et al reported a 50 year old woman who suffered three attacks within 10 months of somnolence and coma lasting a few days.13 They felt that the episodes reflected raised intracranial pressure, possibly caused by a transient blood–brain barrier disturbance.
We found no particular genotypes predisposing to this encephalopathic presentation. Although two members of one family (cases 5 and 6) both had an encephalopathic presentation, other members of the same family did not. Two families had the same mutations (C583T and C622T) but these are the two commonest CADASIL mutations in the British population, each occurring in 10 of the initial 48 index cases. This suggests the presentation is modified by other as yet unknown factors. No triggers were identified; most episodes seemed to follow “normal” migraines.
Conclusions
CADASIL should be considered in the differential diagnosis of an acute encephalopathic illness. Clues to the diagnosis include a history of migraine with aura, a family history of stroke or dementia, and white matter abnormalities on MRI. The encephalopathy is self limiting but may recur.
Acknowledgments
We are grateful to the British Neurological Surveillance Unit and the British Association of Stroke Physicians for assistance in recruiting patients with CADASIL, to all the UK physicians who referred suspected cases, and to Dr G Saldanha who referred case 4. This work was supported by funding received from the NHS Executive London research and development Programme. The views expressed in the publication are those of the authors and not necessarily those of the NHS Executive or the Department of Health.
REFERENCES
Summary of the main clinical features and mutations of six encephalopathic cases of CADASIL
Footnotes
-
Competing interests: none declared