Article Text

Abstract
Background HHT is an autosomal dominant disease with an estimated prevalence of at least 1/5000 which can frequently be complicated by the presence of clinically significant arteriovenous malformations in the brain, lung, gastrointestinal tract and liver. HHT is under-diagnosed and families may be unaware of the available screening and treatment, leading to unnecessary stroke and life-threatening hemorrhage in children and adults.
Objective The goal of this international HHT guidelines process was to develop evidence-informed consensus guidelines regarding the diagnosis of HHT and the prevention of HHT-related complications and treatment of symptomatic disease.
Methods The overall guidelines process was developed using the AGREE framework, using a systematic search strategy and literature retrieval with incorporation of expert evidence in a structured consensus process where published literature was lacking. The Guidelines Working Group included experts (clinical and genetic) from eleven countries, in all aspects of HHT, guidelines methodologists, health care workers, health care administrators, HHT clinic staff, medical trainees, patient advocacy representatives and patients with HHT. The Working Group determined clinically relevant questions during the pre-conference process. The literature search was conducted using the OVID MEDLINE database, from 1966 to October 2006. The Working Group subsequently convened at the Guidelines Conference to partake in a structured consensus process using the evidence tables generated from the systematic searches.
Results The outcome of the conference was the generation of 33 recommendations for the diagnosis and management of HHT, with at least 80% agreement amongst the expert panel for 30 of the 33 recommendations.
- Guidelines
- hereditary hemorrhagic telangiectasia
- arteriovenous malformation
- telangiectasia
- epistaxis
- guidelines
Statistics from Altmetric.com
- Guidelines
- hereditary hemorrhagic telangiectasia
- arteriovenous malformation
- telangiectasia
- epistaxis
- guidelines
Introduction
Hereditary haemorrhagic telangiectasia (HHT) is an autosomal dominant disease with an estimated prevalence of 1/50001 and is thought to be present in all races and parts of the world. Although epistaxis is the most common symptom of HHT and mucocutaneous telangiectasia the most common sign,2 HHT is also often complicated by the presence of arteriovenous malformations (AVMs) in the brain, lung, gastrointestinal (GI) tract and liver.
Unfortunately, HHT is often not diagnosed, and entire families therefore remain unaware of available screening and treatment, and children and adults unnecessarily develop stroke or life-threatening haemorrhage. The goal of the international HHT guidelines process was to develop evidence-based consensus guidelines for the diagnosis of HHT, the prevention of HHT-related complications, and treatment of symptomatic disease.
Methods
The overall guidelines process (figure 1) was developed using the AGREE framework3 with guidelines methodologists. The structure was that of a systematic evidence-based process with incorporation of expert evidence in a structured consensus process where evidence was lacking. We expected only weak or poor evidence in most areas, but chose this approach to maximise quality and applicability of the guidelines and provide a foundation for future research and guidelines in HHT.

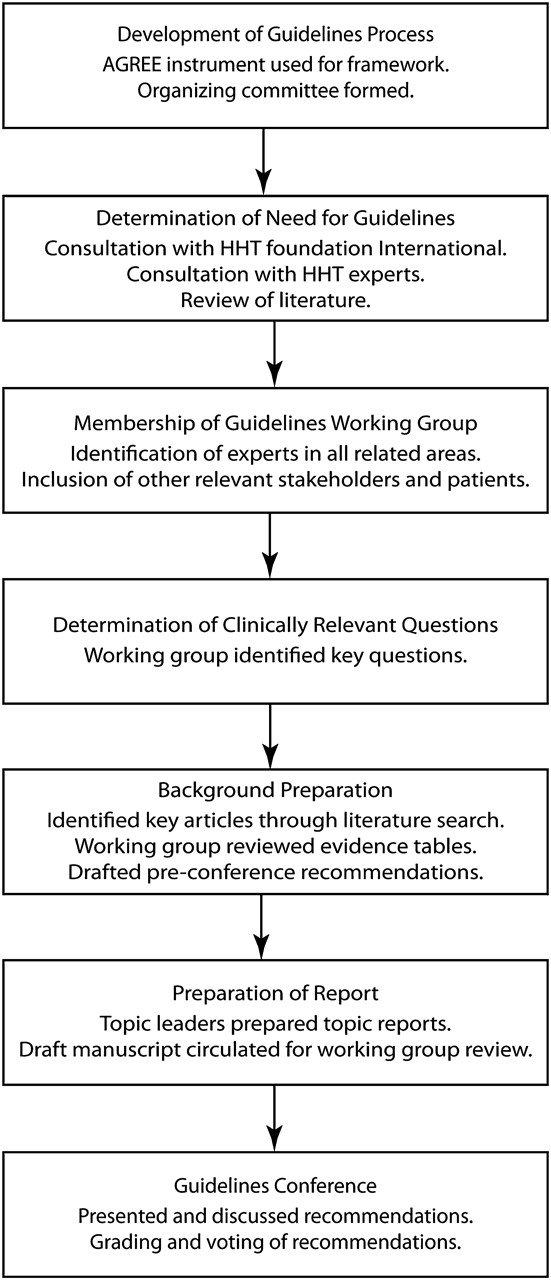{kind=link}
The adopted process of guideline development. HHT, hereditary haemorrhagic telangiectasia.
Determination of need for guidelines
The need for clinical guidelines for HHT was identified by the HHT Foundation International, an international advocacy group for people with HHT, and the Foundation's Scientific and Medical Advisory Board. This was based on their consistent observations of care gaps in HHT, specifically that HHT is underdiagnosed, that there are often delays in diagnosis, and that most patients and families with HHT are not receiving appropriate preventive treatment. No clinical guidelines were in place for the multisystem manifestations of the disorder, except for guidelines for liver vascular malformations (VMs).4
Membership of the HHT guidelines working group
An organising committee of clinicians, scientists, methodologists, patients and Foundation members selected the members of the HHT Guidelines Working Group. This included experts (clinical and genetic) from 11 countries, in all aspects of HHT, guidelines methodologists, healthcare workers and administrators, HHT Foundation representatives, and patients with HHT. Each member was also a member of a topic subgroup (diagnosis, epistaxis, cerebral vascular malformations (CVMs), pulmonary AVMs (PAVMs), GI bleeding and liver VMs). Patients contributed to the development of the clinically relevant questions and the recommendations, with particular input regarding values around recommendations.
Determination of clinically relevant questions
During the pre-conference process, the topic subgroups worked by email to develop clinically relevant questions. The subgroups circulated and edited these through several iterations. These formed the basis for the literature review.
Background preparation
A literature search was conducted using the OVID Medline database from 1966 to October 2006 to identify relevant English-language publications, using the search strategies as outlined in table AI (see online). Hand searches of relevant articles and reviews were also performed for each clinically relevant question. Bibliographies of retrieved publications were reviewed to identify sources not obtained in our search. Publications in abstract form were included to minimise publication bias. One author (MEF) and the literature review assistant (J Silver) independently reviewed abstracts, and any relevant studies were pulled for review. Inclusion and exclusion criteria for study selection are listed in table AI (see online). Results from selected studies were extracted into evidence tables, and, along with original papers, were sent to participants for review and to determine if any relevant literature was missing.
Determination of clinical recommendations
Participants convened at the Guidelines Conference to take part in a structured consensus process using the evidence tables. With the assistance of professional guidelines facilitators, topic subgroups prioritised clinically relevant questions and then generated recommendations for these. All participants assembled afterwards to vote for all generated recommendations. Those recommendations achieving less than 80% agreement were further discussed, revised again with a facilitator, and voted on again. Wording of recommendations was considered final, and they are presented with the percentage agreement obtained on the final vote. Priorities for future research were also identified during the process (see online table AII).
Grading of evidence
Each recommendation was graded to indicate the level of evidence available using the classification system of the Canadian Task Force on the Periodic Health Examination5 (table 1). In addition, values around recommendations were generated using the GRADE instrument,6 7 and these were reported as ‘strength of the recommendation’. The ‘strength of the recommendation’ incorporated evidentiary and non-evidentiary factors, including baseline risks of outcomes, benefits of treatment, potential harms of treatment, certainty of point estimates, and levels of evidence. Values were also incorporated, such as the importance of certain outcomes to stakeholders and other factors such as availability of certain tests.
Categorisation of the quality of evidence
General organisation
The pre-conference process occurred by email over 6 months leading up to the 2-day Guidelines Conference near Toronto, Canada, in November 2006. The Conference was held in a facility with anonymous key pad voting technology. The large group sessions were recorded (audio) and minuted.
Preparation of report
Topic leaders generated each area of this article, which was then revised by MEF, VP and the topic members for each group, and then reviewed by the other authors. The literature search referenced was that obtained in October 2006. At the time of final manuscript review, two steps were taken to ensure that no generated recommendation needed immediate revision. Firstly, a literature search for any interim randomised controlled trials in HHT was performed, which revealed none. Secondly, the Working Group was polled for knowledge of any recent publications that would lead to a significant change in any of the recommendations, and none were identified.
Role of funding sources
Although the funding organisations were not directly involved in the generation of the recommendations, some of the participants in the guidelines process were also board members of the HHT Foundation International and its Scientific and Medical Advisory Board. The other funding sources had no role in the design, conduct and reporting of the study or in the decision to submit the results for publication.
Diagnosis of HHT
Background
Making the diagnosis of HHT in a patient allows the appropriate screening and preventive treatment to be undertaken in the patient and their affected family members. HHT has traditionally been diagnosed on the basis of its clinical features, but can now also be diagnosed using genetic testing. We reviewed the evidence and expert experience for clinical and genetic diagnosis in HHT.
The clinical diagnostic features of HHT have been identified by describing the clinical presentation of patients who have known or suspected HHT and their close relatives. The average age of onset for epistaxis is 12 years, with nearly 100% affected by age 40 years.2 8–10 Most patients report the appearance of telangiectasia of the mouth, face or hands 5–30 years after the onset of nose bleeds, most commonly during the third decade. Unfortunately, there are no longitudinal natural history studies of HHT clinical manifestations and how these might vary with genotype.
In 2000, consensus clinical diagnostic criteria known as the Curaçao Criteria were published11 (table 2). Using these criteria, a diagnosis of HHT is considered ‘definite’ if three or more criteria are present, ‘possible or suspected’ if two criteria are present, and ‘unlikely’ if 0 or 1 criterion is present.
Curaçao Criteria for clinical diagnosis of hereditary haemorrhagic telangiectasia (HHT)
There have been no studies reporting sensitivity and specificity of the Curaçao Criteria, but the expert panel agreed that the Curaçao Criteria are particularly helpful in two situations: (1) discriminating affected from non-affected older adults and (2) ruling-in the diagnosis in younger adults and children. The expert panel was specifically concerned about the risk of missing diagnoses in children and young adults, who might have no epistaxis or visible telangiectases, yet have undiagnosed PAVMs or CVMs.12 It is in these groups that genetic testing should be most useful.
The goal of genetic testing for HHT is to clarify the specific HHT mutation in an HHT family, allowing diagnosis among those relatives (often children and young adults) who do not meet clinical diagnostic criteria. Genetic testing is performed first on the index case in the family and involves DNA sequencing and deletion/duplication analysis of the coding exons of the endoglin gene (ENG, HHT1) and the activin A receptor type II-like 1 gene (ACVRL1, HHT2). Mutations in these genes account for the majority of cases of HHT. At least two other HHT loci have been described, although specific genes at these loci are not yet identified.13 14 Mutations in the SMAD4 gene can cause a rare syndrome which combines juvenile polyposis and HHT.15 Genetic testing in HHT is complex relative to many other genetic conditions because a mutation in one of multiple genes can cause the condition, not all genes that can cause HHT have been discovered, and there are no ‘common mutations’, with most families having their own ‘private’ HHT mutation.
Several authors have reported16 17 a clinical sensitivity/mutation detection rate of ∼75% for sequence analysis of ENG and ACVRL1. Use of an additional method to detect large deletion/duplication mutations increases the detection rate by ∼10%.16 17 Recent reports suggest that about 1–3% of patients clinically diagnosed with HHT will have a mutation detected in the SMAD4 gene, or about 10% of those who test negative for ENG and ACVRL1 mutations.17–19
There is considerable clinical overlap between patients/families with ENG mutation and those with ACVRL1 mutation, with VMs reported in similar organs in both types.20–22 The expert panel agreed that ENG versus ACVRL1 genotype should not significantly influence screening recommendations for VMs. Most HHT patients/families with SMAD4 mutation reported to date have juvenile polyposis and are therefore at risk of GI malignancy.15 18
There is currently no evidence about the effect of prenatal testing for HHT and no consensus among experts about how fetal diagnosis might alter pregnancy or delivery management. Expert experience is that prenatal diagnosis is not commonly sought in HHT, and is most often requested as an alternative to postnatal diagnostic testing when there is already another reason for performing prenatal testing.
Recommendations
|
|
|
|
|
|
The expert panel recommends that clinicians refer patients for diagnostic genetic testing for HHT
|
|
|
|
|
|
Epistaxis
Background
Recurrent spontaneous epistaxis is the most common symptom of HHT and often leads to iron-deficiency anaemia.24 Epistaxis appears before the age of 20 years in about 50% of patients, with 78–96% of all HHT patients developing it eventually.2 During the guidelines development process, patients identified epistaxis as a priority HHT-related health concern affecting their everyday life, and the literature suggests that epistaxis is an important factor reducing quality of life in HHT.25 We reviewed the evidence for treatment of HHT-related epistaxis, searching for studies regarding treatment of the usual chronic recurrent epistaxis as well as of acute episodes of epistaxis requiring urgent medical consultation.
Non-invasive management of chronic recurrent epistaxis in HHT has focused to date on prevention of epistaxis events through measures to maintain integrity of the nasal mucosa, such as humidification. The rationale for humidification is that endonasal crusting and airflow lead to damage of endonasal telangiectasia and secondary bleeding, whereas humidification should help prevent endonasal crusting. There are small case series of various topical medications, including lubricants (eg, saline, antibiotic ointments),26 27 as well as topical oestrogen cream/ointment28 and antifibrinolytics,29 with variable success in decreasing HHT-related epistaxis. There are insufficient published data to recommend one topical therapy over another; however, expert experience is that there is mild benefit from humidification and that the risk of topical lubricants and saline is very low.
Procedural therapies for chronic HHT-related epistaxis include endonasal laser, electrical or chemical coagulation techniques, replacement of the fragile endonasal mucosa by skin or buccal mucosa (dermoplasty), nasal artery embolisation and closure of the nasal cavity (known as Young's procedure). There have been no controlled or well-designed comparative studies of any of these procedures in HHT-related epistaxis, for either acute or chronic management. Case series and expert opinion of endonasal coagulation for treatment of moderate HHT-related epistaxis suggests that most types of endonasal coagulation appear to be low-risk procedures with subjective improvement in most patients.24 26 30–34 Chemical cautery (eg, AgNO3) and CO2 laser coagulation appear to have poorer outcomes in HHT and higher risk of intraoperative bleeding.26 Septal dermoplasty has been reported, in one uncontrolled retrospective case series of patients with severe epistaxis, to decrease mean transfusion requirements and to improve subjective quality of life, but follow-up was available in <50% of treated patients35 and complications included endonasal crusting and dryness. Young's procedure has been shown in a few small case series of patients with severe epistaxis to cause cessation of epistaxis and also to improve quality of life, although patients report side effects of chronic mouth breathing.36–38 Nasal artery embolisation is generally not useful for treatment of chronic epistaxis, since the effect is generally short term.39 40 Submucosal or intravascular endonasal injections of different substances have been reported,41 42 often with reduction in epistaxis but also reports of complications such as severe allergic reactions and blindness.42
The expert panel agreed that, given the learning curve for surgical management of chronic HHT-related epistaxis, involvement of surgeons with expertise in HHT-related epistaxis may increase the likelihood of appropriate choice of treatment and improve outcomes of therapy. The expert panel also agreed that this applied to nasal surgery for indications other than epistaxis, in HHT patients.
Several medical treatments have been reported for HHT-related epistaxis, but there are no well-designed studies supporting their effectiveness and most studies have been limited by the lack of a validated sensitive outcome measure. There is one negative randomised placebo-controlled double-blind trial of oestrogen,43 and another of tranexamic acid,44 in which investigators were unable to demonstrate significant improvement in haemoglobin (primary outcome) but did demonstrate significant improvement in subjective epistaxis (secondary outcome).44
There are no well-designed studies of the first-line management of acute epistaxis, although nasal packing is often used to control acute bleeding. However, endonasal telangiectasias are extremely fragile and therefore packing removal can cause rebleeding. This can be minimised with atraumatic packing—for example, using lubricated or pneumatic packing, the latter allowing insertion and removal of the packing in a deflated size. Low-pressure pneumatic packing may also minimise mucosal ischaemic damage. Two uncontrolled case series of embolisation,40 45 in patients with severe ongoing epistaxis despite packing, reported excellent immediate success rates (80–100%), but with early recurrence of epistaxis and risk of serious procedural complications (stroke, tissue necrosis).
The panel also discussed management when an HHT patient has an indication for antiplatelet or anticoagulant therapy. There are no published studies regarding the use of anticoagulants in HHT, but expert experience revealed a wide range of outcomes, with some HHT patients tolerating anticoagulation and others developing life-threatening bleeding.
Recommendations
| The expert panel recommends that physicians advise patients with HHT-related epistaxis to use agents that humidify the nasal mucosa to prevent epistaxis. |
|
|
|
|
|
|
|
|
|
|
|
Cerebral vascular malformations
Definition
The term CVM refers to a variety of vascular abnormalities, classified on the basis of morphology, including: (1) arteriovenous malformations (CAVMs) (including microAVMs measuring <1 cm in size); (2) cavernous malformations; (3) venous angiomas/developmental venous anomalies (DVAs); (4) capillary telangiectasias, enlarged capillary-sized vessels; (5) vein of Galen malformations; (6) high-flow pial fistulae (arterivenous fistulae (AVFs)); and (7) mixed malformations.46 All of these types of CVMs can be found in HHT patients, although typically HHT is associated with CAVMs, AVFs, microAVMs and telangiectasias.47
Background
Approximately 23% of HHT patients will harbour a CVM.48–50 The rationale for screening for CVMs in HHT is that screening will detect a treatable CVM before the development of a life-threatening or debilitating complication. We therefore reviewed the evidence regarding complications of CVMs, the performance of screening tests, and the effectiveness of treatment for CVMs. Given the rarity of HHT-related CVMs, most of the evidence reviewed relates to the more common sporadic CVMs.
The bleeding risk of CVMs in HHT has been estimated retrospectively at ∼0.5% per year,51 although there are no prospective natural history studies. In larger series of sporadic CAVMs,52 the annual rate of rupture is 2–4%/year.52 On the basis of case series, CAVMs and AVFs appear to have a more aggressive natural history, while cavernous malformations (CM), capillary telangiectasias and DVAs, also reported to occur in HHT,49 appear to have a more benign natural history.12 48 51 53 54 There are several case series and reports of catastrophic haemorrhagic sequelae of CVMs and spinal AVFs occurring during childhood.50 55–58 Rarely, spontaneous resolution of CVMs has been reported.59 60
The typical imaging features of HHT CVMs include the presence of either multiple, cortical, micro AVMs or AVFs harbouring single feeding arteries and single draining veins.12 53 54 Catheter angiography remains the ‘gold standard’ for diagnosis of most types of CVMs, but carries a 0.5% risk of permanent stroke.61 MRI is considered to be a safe, non-invasive modality to screen for CVMs, but unfortunately there are no screening studies assessing its performance in HHT. MRI screening studies for non-HHT CVMs have been limited by small size, retrospective design and lack of blinding to clinical status, but suggest sensitivity of 80–95% for medium to large sized CVMs.62–64 MRI is less sensitive for the detection of micro AVMs,64 but the addition of contrast enhancement (gadolinium for patients >2 years of age) to MRI sequences increases the sensitivity for microAVMs. The inclusion of sequences designed to detect blood products (currently gradient echo sequences) also increases the sensitivity of MRI for microAVMs and signs of asymptomatic haemorrhage.55 ‘False-positive’ results may occur when other types of CVMs are encountered including telangiectasias which have a favourable natural history49 and for which no further invasive imaging is required. Transcranial Doppler ultrasonography (US) has also been used to screen for CVMs,65 66 with reported sensitivity of ∼80% for medium to large-sized CVMs, but studies are limited by sample size and design. No evidence exists for follow-up screening after an initial negative study, as there is no evidence to suggest that adult patients with HHT develop new CVMs.
MRI provides a relatively safe, sensitive testing modality to identify CVMs in children.67 While MRI itself poses little risk, the expert panel acknowledges the risk related to sedation/anaesthesia of children for diagnostic procedures. Of greatest concern is the risk of respiratory depression, but this should be minimised with appropriate cardiorespiratory monitoring. No evidence exists at this time to recommend follow-up screening after an initial negative study during childhood, but consideration should be given to one adulthood MRI following initial negative childhood MRI.
The expert panel agreed that CVM obliteration is required to effectively eliminate the future risk of haemorrhage. Although treatment may provide a large relative risk (RR) reduction for cerebral bleeding, procedural risks are significant. There are no published studies of the efficacy or safety of any form of treatment of CVMs in HHT patients. However, several large case series (>200 patients, mostly single-centre) of embolisation, microsurgery and stereotactic radiation in non-HHT CAVMs show widely ranging effectiveness for each modality.49 54 56 68–78 On the basis of this, as well as expert experience, the expert panel agreed that effective treatment strategies include embolisation, microsurgery and stereotactic radiation, or combinations of these. With the rarity of CVMs and the associated risks of treatment, the expert panel agreed that each case should be managed in an individualised manner and decisions about invasive testing and therapy should occur at centres with significant experience and expertise in all treatment modalities. Although there is no evidence regarding differences in outcomes according to expertise in management of these cases, the expert panel agreed that centres with experience in HHT-related CVMs will be more aware of important issues related to the care of HHT patients and likely to have better outcomes of surgical and other procedures.
CVMs occur in infants and children with HHT.12 47 50 57 79 80 Before the age of 6 these malformations tend to be high-flow pial fistulae (cerebral or spinal cord AVFs).47 Expert opinion is that these malformations have a more aggressive natural history than nidus-type CAVMs, including presenting events such as intracerebral haemorrhage, cognitive deficit, cardiac insufficiency, epilepsy and hydrocephalus.12 47 79 80 Embolisation or microsurgical obliteration of these high-flow pial fistulae in children may therefore be of significant benefit when performed by a neurovascular centre with expertise in these techniques in children.
There is no evidence to guide the management of CVMs during pregnancy and delivery, as there is no good evidence regarding the risk of CVM complications or treatment during pregnancy and delivery.
Recommendations
|
|
|
|
|
|
| The expert panel recommends that adults presenting with an acute haemorrhage secondary to a CVM be considered for definitive treatment in a centre with neurovascular expertise. |
|
|
|
|
|
Pulmonary arteriovenous malformations
Background
PAVMs are present in 15–50% of people with HHT and have been associated with life-threatening complications, as previously reviewed.81 82 The rationale for screening HHT patients for PAVMs is that screening will detect a treatable PAVM before the development of a life-threatening or debilitating complication. We therefore reviewed the evidence regarding complications of PAVMs, the performance of screening tests, and the effectiveness of treatment for PAVMs.
PAVMs have been shown to be associated with disabling and life-threatening complications, such as stroke, transient ischaemic attack (TIA), cerebral abscess, massive haemoptysis and spontaneous haemothorax81 83–86 in retrospective series. The neurological complications are presumed to occur via paradoxical embolisation through PAVMs, whereas the haemorrhagic complications occur due to spontaneous PAVM rupture. These complications have been demonstrated in largely adult series of HHT patients, although they have also been demonstrated in a paediatric HHT series,87 albeit smaller in size. There have also been small series reporting these same complications during pregnancy,88 89and the expert panel agreed that the complication risk appears to be greater during pregnancy.
Since clinical symptoms and signs of PAVMs are often absent before the development of complications, a number of screening tests have been studied, including physiological methods of measurement of intrapulmonary shunt as well as multiple different imaging modalities. In the one comparative study (table 3), transthoracic contrast echocardiography with agitated saline (TTCE) has been demonstrated to have the best combination of high sensitivity82 and low risk90 91 among screening tests for PAVMs in adults with HHT, when compared with the reference standard tests (CT and pulmonary angiography). There have been no comparative screening studies for PAVMs in children with HHT.
Level II study of screening tests for pulmonary arteriovenous malformations (PAVMs) in patients with hereditary haemorrhagic telangiectasia (HHT), using reference standard
Embolisation has been shown in several non-controlled series83 85 92–96 to be efficacious and to have a good safety profile, with only rare PAVM-related complications during 5–10 year follow-up (table 4) . In the short term, these studies showed very high rates of immediate technical success and significant improvement in oxygenation (table 4). Longer term after embolisation, reperfusion did occur in up to 15%, and growth of small PAVMs in up to 18% (table 4), but clinical complications were very rare. These series primarily reported outcomes for treatment of PAVMs with feeding artery diameter of 3 mm or greater, although expert experience suggests that embolisation of smaller PAVMs (2–3 mm) has similar outcomes. The safety and efficacy were similar for large PAVMs in adults97 as well as for PAVMs in children,87 although there is little experience with embolisation of PAVMs in children under the age of 4 years. There is only one small case series of embolisation during pregnancy,98 suggesting reasonable safety. Although there is no evidence regarding differences in outcomes according to expertise in embolisation of PAVMs, the expert panel agreed that centres with experience in this procedure are more likely to have better outcomes than inexperienced centres.
Level II uncontrolled case series of transcatheter embolisation (detachable coils, balloons, etc) for pulmonary arteriovenous malformations (PAVMs)
The long-term follow-up of PAVMs is described using CT of the thorax. This allows detection of reperfusion by non-involution of the aneurysmal sac ∼1 year after embolisation and also detection of growth of small residual PAVMs, which are common in HHT.85 TTCE has been shown to be not useful after embolisation, given that it remains positive in ∼90% of patients after embolisation.99
Recommendations
|
|
|
|
|
|
|
|
|
|
Gastrointestinal bleeding
Background
Although 80% of patients with HHT have gastric or small intestinal telangiectasia100 on endoscopy or capsule examination, only 25–30% of patients will develop symptomatic GI bleeding,1 2 101 102 which usually does not present until the 5th or 6th decades of life. Patients rarely develop significant GI bleeding before 40 years of age.1 2 101 102 Women are affected with GI bleeding in a ratio of 2–3:1.103 104
Patients with HHT and GI bleeding may or may not be symptomatic, as the bleeding is usually in a slow, chronic and intermittent fashion, often without notable melaena. Patients often have few symptoms until they become anaemic. In severe cases, HHT GI bleeding causes morbidity, dependency on blood transfusions, and increased mortality.103 Severity of GI bleeding in HHT is generally based on severity of the anaemia. Gastric and duodenal telangiectasias are more common than colonic telangiectasias and contribute more to overall GI bleeding and chronic anaemia in HHT patients.105
At present, endoscopic evaluation is considered the gold standard test for evaluation of GI bleeding in HHT patients. Although the majority of patients with HHT will have GI telangiectasias, the utility of endoscopic evaluation is in the anaemic or iron-deficient patient. The presence and number of gastric and duodenal telangiectasias have been shown to predict the presence and number of jejunal telangiectasias,104 and therefore, for diagnostic purposes, an oesophagogastroduodenoscopy is sufficient in most cases.
Management of GI bleeding in HHT involves treatment of the iron-deficiency/anaemia and therapies to reduce GI bleeding. Treatment of anaemia and iron deficiency includes aggressive iron replacement and blood transfusions as necessary. There are no studies of iron replacement in HHT, but experts agree that oral iron supplementation may be sufficient in some patients, although consideration of intravenous iron supplementation may be necessary in more severe cases. There have been no studies of erythropoietin therapy in HHT, but it is sometimes considered in severe cases, in combination with iron, in an attempt to accelerate treatment of the anaemia.
Current treatment options to reduce chronic GI bleeding in HHT include hormonal therapy (oestrogen/progesterone preparations or danacrine), antifibrinolytics (aminocaproic acid or tranexamic acid), other medications reported only as isolated case reports (tamoxifen, interferon, thalidomide and sirolimus) and endoscopic therapy. There is one small double-blind placebo-controlled cross-over trial106 (table 5) of combination hormonal therapy (ethinylestradiol 0.05 mg plus norethisterone 1 mg) versus placebo, each for 6 months, in 10 patients with transfusion-dependent severe GI bleeding. Five of the six HHT patients had no further GI bleeding while receiving treatment, and, in the overall group, there was a significant decline in transfusion requirements. In a retrospective case series103 of 43 HHT patients with GI bleeding, median haemoglobin improved significantly (8.6 to 9.8 mg/dL, p=0.0018) for the 23 patients treated with medical therapy (ethinylestradiol/norethindrone in 19, danacrine in two, and aminocaproic acid in two). Although there are only other individual case reports107 of danacrine in HHT GI bleeding, it may be a reasonable alternative to oestrogen/progesterone therapy in male patients, as it does not have feminising effects. There is only individual case report evidence for antifibrinolytics for HHT-related GI bleeding,108 but there is expert experience suggesting benefit in these patients. Overall, there is insufficient evidence to recommend any medical treatment as first-line therapy in these patients, given the potential side effects; however, there may be a role for these agents when iron replacement is insufficient to control anaemia.
Therapeutic trials for gastrointestinal (GI) bleeding in hereditary haemorrhagic telangiectasia (HHT)
There are small case series (table 5) and expert experience suggesting that local endoscopic therapy, using argon plasma coagulation (APC) or ND-YAG laser, may be beneficial in reduction of HHT-related GI bleeding. In three small case series109–111 of repeated ND-YAG therapy, transfusion requirements declined in more than 50% of patients. The expert panel agreed that, although the reported series were primarily of the use of ND-YAG laser, APC is the most effective method of endoscopic therapy currently available. Overall, there is insufficient evidence to recommend endoscopic therapy as first-line therapy in HHT-related GI bleeding; however, there may be a role for endoscopic therapy when iron replacement is insufficient to control anaemia. There is no evidence or experience supporting cauterisation of colonic telangiectasia, or for surgery or transcatheter embolotherapy in the routine management of HHT-related GI bleeding. Although there is no evidence regarding differences in outcomes according to expertise in endoscopic management of GI bleeding in HHT, the expert panel agreed that clinicians with experience in HHT-related GI bleeding will better prepared to make decisions about when to treat GI telangiectasia in HHT and are likely to achieve better outcomes for these procedures.
There is no evidence of any benefit from altering nutrition or lifestyle, or screening for Helicobacter pylori in patients with HHT-related GI bleeding. HHT patients with GI bleeding should avoid anticoagulants and drugs that alter platelet function. However, when other comorbidities require use of these drugs, expert experience is that these can often be tolerated, especially when doses are kept as low as possible.
Recommendations
|
|
|
|
|
|
|
|
|
|
Liver vascular malformations
Background
Although a consensus guideline had been recently published for the diagnosis and management of liver VMs in HHT,4 to be consistent, we elected to include this topic in the present guidelines. As such, we followed the same guidelines process for liver VMs as for other aspects of HHT and reviewed the evidence regarding diagnosis and treatment of liver VMs in HHT. The liver VMs recommendations reported in the present guidelines do not differ significantly from the previous guidelines.4
Liver VMs are present in 32–78% of HHT patients112–116 (table 6). Although there are no published natural history data regarding liver VMs in HHT, it appears that symptoms occur in only about 8% of the patients with HHT and liver VMs.114 117 The clinical presentations of liver VMs include high-output heart failure, portal hypertension and biliary necrosis.118
Screening studies for liver vascular malformations (VMs) in hereditary haemorrhagic telangiectasia (HHT)
In patients who have symptoms suggestive of liver VMs,118 it is important to establish the diagnosis of liver VMs for therapeutic and prognostic purposes. The diagnosis of liver VMs may also assist in the clinical diagnosis of HHT, since visceral involvement is one of the clinical diagnostic criteria.11 Several different imaging modalities have been reported and studied for the screening and diagnosis of liver VMs in HHT. From the least invasive to the most invasive, these tests are Doppler ultrasonography, MRI, triphasic spiral CT and mesenteric angiography. Doppler US is the least invasive test, requiring no contrast and being associated with no procedural complications. There is little experience with MRI, which does require MR-contrast administration, but involves no radiation exposure. CT is associated with radiation exposure and risk of contrast allergy. Mesenteric catheter angiography has traditionally been considered the diagnostic gold standard, but is the most invasive, and is rarely used.
Typical angiographic findings have been described in several small case series of HHT patients,119–121 including telangiectasias, confluent VMs, hepatic artery dilatation and shunting (arterioportal, arteriovenous and/or portovenous). Triphasic CT findings have been similarly described.112 113 117 Several case series of Doppler US in HHT patients have demonstrated hepatic artery dilatation, elevated hepatic artery flow, and intrahepatic hypervascularity.113 114 116 122 123 There have been no well-designed studies reporting sensitivity and specificity of any of these tests, although the positive predictive value of Doppler US appears to be near 100%.115 123 Screening studies of HHT patients (table 6) have reported a prevalence of liver VMs of 32–72% with Doppler US114–116 and 67–78% with triphasic CT.112 113 In none of these studies was a diagnostic gold standard (angiography) uniformly performed; however, these prevalences are all much higher than the symptomatic rate (8%), suggesting that these tests are sensitive. There are no screening studies in children.
Histological diagnosis from liver biopsy tissue, although quite characteristic,118 is unnecessary, given typical imaging findings, and risky in patients with liver VMs. Focal nodular hyperplasia occurs more often in HHT than in the general population,124 but can be diagnosed through imaging, without biopsy.
There are three uncontrolled case series (table 7) of treatments of liver VMs, specifically hepatic artery embolisation and liver transplantation. Hepatic artery embolisation has the objective of reducing arteriovenous or arterioportal shunting by embolising branches of the hepatic artery. Embolisation appears to be effective in improving symptoms related to high-output heart failure and mesenteric steal syndrome125; however, the effect is transient and symptoms generally recur. More importantly, ischaemic complications (ischaemic cholangitis, ischaemic cholecystitis and/or hepatic necrosis) leading to transplant or death occur in ∼30% of the treated cases, including 50% of treated portal hypertension cases.125 The 2-year survival with embolisation was ∼73%. The expert panel agreed that the risk of post-embolisation ischaemia would probably be greatest in patients with biliary presentation of liver VMs. With liver transplantation, symptoms resolved in the majority of patients.126 127 Liver transplantation is associated with high blood transfusion requirements, prolonged hospital stay, and a relatively high rate of postoperative complications. However, the reported 5-year survival rate of 83% in the larger series127 compared favourably with overall survival rates for liver transplantation.
Case series of treatment for liver vascular malformations (VMs) in hereditary haemorrhagic telangiectasia (HHT)
Recommendations
|
|
|
|
|
|
|
|
|
|
Acknowledgments
Many thanks to the following people who contributed at different stages to the HHT Guidelines: Marianne Clancy MPA, Justine Cohen-Silver MD MSc, Sharon Straus MD MSc, Dave Davis MD, Mary Ellyn Parker RN, Ana Carvalho, Joyce Fenuta RN BScN MHS, Beth Plahn RN MHA, David Stoner BS RPh, Keith Williams, Cheryl Wilson MA MS, Jane E Tumpson MAT MBA, Samir Gupta MD MSc, Kevin Sanders MD MSc, Anna R. Gagliardi PhD, Andrew Worster MD and Denice Feig MD MSc.
References
Footnotes
The HHT Guidelines Working Group intends to generate updated clinical guidelines within approximately 5 years.
Centres with recognised expertise in the diagnosis and management of HHT can be located at http://www.hht.org/, the website for the HHT Foundation International.
Funding John Abele on behalf of the Argosy Foundation, HHT Foundation International Inc, Canadian Institutes of Health Research, St Michael's Hospital Department of Medicine. Financial support for MEF: Nelson Arthur Hyland Foundation, Li Ka Shing Knowledge Institute of St Michael's Hospital.
Competing interests VP received an honorarium for attending the HHT Guidelines Conference. DP received a grant (completed) from the Ethek F Donahue Foundation to study hormonal therapy in HHT.
Provenance and peer review Not commissioned; externally peer reviewed.