Article Text

Abstract
Background: The most commonly reported phenotypes described in patients with PTEN mutations are Bannayan–Riley–Ruvalcaba syndrome (BRRS), with childhood onset, macrocephaly, lipomas and developmental delay, and Cowden Syndrome (CS), an adult-onset condition recognised by mucocutaneous signs, with a risk of cancers, in particular those of the thyroid and breast. It has been suggested that BRRS and CS are the same condition, but the literature continues to separate them and seek a genotype–phenotype correlation.
Objective: To study the clinical features of patients with known PTEN mutations and observe any genotype–phenotype correlation.
Methods: In total, 42 people (25 probands and 17 non-probands) from 26 families of all ages with PTEN mutations were recruited through the UK clinical genetics services. A full clinical history and examination were undertaken.
Results: We were unable to demonstrate a genotype–phenotype correlation. Furthermore, our findings in a 31-year-old woman with CS and an exon 1 deletion refutes previous reports that whole exon deletions are only found in patients with a BRRS phenotype.
Conclusion: Careful phenotyping gives further support for the suggestion that BRRS and CS are actually one condition, presenting variably at different ages, as in other tumour-suppressor disorders such as neurofibromatosis type 1. This has important counselling implications, such as advice about cancer surveillance, for children diagnosed with BRRS.
- BRRS, Bannayan–Riley–Ruvalcaba syndrome
- CS, Cowden syndrome
- VATER, vertebral defects, anal atresia, trachea-oesophageal fistula with oesophageal atresia, radial and renal abnormalities
- PTEN
- Bannayan-Riley-Ruvalcaba syndrome
- Cowden Syndrome
- genotype-phenotype correlation
- variable expressivity
Statistics from Altmetric.com
- BRRS, Bannayan–Riley–Ruvalcaba syndrome
- CS, Cowden syndrome
- VATER, vertebral defects, anal atresia, trachea-oesophageal fistula with oesophageal atresia, radial and renal abnormalities
- PTEN
- Bannayan-Riley-Ruvalcaba syndrome
- Cowden Syndrome
- genotype-phenotype correlation
- variable expressivity
The PTEN gene is a constitutively active tumour suppressor gene encoding a phosphatase with lipid and protein substrates, active in many pathways involved in cellular growth. Its role in the phosphoinositol 3-kinase pathway, which links several hamartoma syndromes including tuberous sclerosis and Peutz–Jeghers syndrome, is well studied.
Cowden syndrome (CS) and Bannayan–Riley–Ruvalcaba syndrome (BRRS) are the most commonly reported conditions caused by mutations in the PTEN gene.1–7 Both are characterised by multiple hamartomas and have many overlapping features. The diagnostic criteria for CS8,9 (box 1) are based on the presence of pathognomic mucocutaneous lesions including trichilemmomas, acral keratoses and mucosal neuromas and Lhermitte–Duclos syndrome, a rare hamartomatous dysplastic gangliocytoma of the cerebellum.10–17 Cutaneous manifestations are reported to be fully penetrant by the second decade.10 CS is rarely found before adulthood. There is a predisposition to malignancy, particularly cancers of the thyroid (lifetime risk 3–10%) and breast (lifetime risk 25–50%).10,18–20 The frequency of endometrial cancer is unknown.21 Other common CS features include goitre, thyroid adenomata, uterine fibroids and fibrocystic disease of the breast. Hamartomatous gastrointestinal polyps are reported, mostly colorectal, although stomach and duodenal polyps are also reported. Colorectal cancers are not a feature of CS.22PTEN mutations have been found in 13–81%2,4,23,24 of people who met the diagnostic criteria for CS. Surveillance as recommended by the National Comprehensive Cancer Network (www.nccn.org; V.1.2007) is “Category 2A....based on NCCN consensus, based on lower level evidence including clinical experience” and the original International Cowden Consortium recommendations.25
Box 1 Cowden Consortium Criteria V.1.2007. From the NCCN website (www.nccn.org)
Pathognomonic criteria
-
Lhermitte–Duclos disease, defined as presence of a cerebellar dysplastic gangliocytoma
-
Mucocutaneous lesions
-
Acral keratoses
-
Trichilemmomas, facial
-
Papillomatous lesions
-
Mucosal lesions
Major criteria
-
Breast cancer
-
Thyroid cancer, especially follicular thyroid cancer
-
Macrocephaly (occipital frontal circumference >97th centile)
-
Endometrial carcinoma
-
Minor criteria
-
Other thyroid lesions (eg, goitre)
-
Hamartomatous intestinal polyps
-
Fibrocystic disease of the breast
-
Mental retardation (IQ <75)
-
Lipomas
-
Fibromas
-
Genitourinary tumors (especially renal cell carcinoma)
-
Genitourinary structural manifestations
-
Uterine fibroids
An operational diagnosis of Cowden syndrome is made if a person meets any one of the following criteria:
-
Pathognomonic mucocutaneous lesions
alone if there are: six or more facial papules, of which three or more must be trichilemmoma, OR
cutaneous facial papules and oral mucosal papillomatosis, OR
oral mucosal papillomatosis and acral keratoses, OR
six or more palmoplantar keratoses
-
Two major criteria but one must be either macrocephaly or Lhermitte–Duclos disease
-
One major and three minor criteria
-
Four minor criteria
In a family in which one member meets the diagnostic criteria for CS, other relatives are considered to have a diagnosis of CS if they meet any of the following criteria:
-
A pathognomonic mucocutaneous lesion
-
Any one major criterion with or without minor criteria
-
History of Bannayan–Riley–Ruvalcaba syndrome
Adapted from Eng.40
There are no agreed international criteria for the diagnosis of BRRS, but Marsh et al26 defined this as at least three of the following four features: macrocephaly, lipomatosis, haemangiomas and speckled penis in males, whereas Parisi et al defined it as at least two of three features of macrocephaly, hamartomas (including at least one lipoma, haemangioma or intestinal polyp) and penile macules in males. BRRS is generally reported with a childhood onset, often with delayed motor and intellectual development. Other features described include thyroid adenomas, Hashimoto’s thyroiditis, lymphatic malformations, joint hyperextensibility, seizures, scoliosis, lipid storage myopathy and a high arched palate. PTEN mutations are reported in 57–60% of BRRS cases.24,27
Other conditions described with PTEN mutations include presentation with macrocephaly or macrosomia and developmental delay without hamartoma or penile macules, resembling older descriptions of benign familial macrocephaly,27,28 autism with macrocephaly,29,30 a VATER-like phenotype31 and a severe phenotype reminiscent of Proteus syndrome.10,32–38
Most studies have failed to demonstrate a consistent genotype–phenotype relationship, and initial suggestions that CS was associated with exon 5 mutations in the PTPase core motif have not been substantiated.2 However, Zhou et al found that promoter mutations were associated with CS and that deletions of the gene or single exon deletions were associated with BRRS.39 Other patients with cytogenetically visible deletions and BRRS have been reported.3,41 More recently, patients with a deletion encompassing BMPR1A and PTEN with juvenile polyposis of infancy were reported. Those patients had macrocephaly, but none of the other features of BRRS.42 A genotype–phenotype correlation with distinct splice-variant profiles resulting in differing predicted downstream effects has also been described.43,44
There is continued debate as to whether all described PTEN-related syndromes are in fact one entity45 and should therefore receive equal attention with respect to cancer surveillance.46–49 In this paper, we present the findings of our clinical study pertinent to this debate and attempt to provide clarification.
METHODS
Ethics approval for the study was obtained from the Southampton and Southwest Hampshire Ethics Committee (MREC 04/Q1702/86). People of any age with a known PTEN mutation were eligible to take part. Participants were recruited through UK clinical genetics services. All patients were visited at home by a researcher (KL). A full history and clinical examination was performed using a standard protocol created for the study. Photographs of patients were taken and reviewed with a senior clinician (IKT). To ensure accuracy of information, molecular and histological reports and clinical details were requested from the participants’ doctors.
RESULTS
In total, 42 mutation-positive people from 26 families were recruited (table 1). Of these, 25 were probands, of whom 15 were <18 years of age, and 17 were non-probands, of whom 3 were<18 years of age. There were 18 male and 7 female probands; the ratio of male:female non-probands was more even (8M:9F). Participants were identified by two numbers: the first is the family number, the second the individual number (1 if a proband; 2,3 etc if a non-proband). In family 26, the proband was not seen.
Clinical features
In 9 of the 26 families in this study, at least 2 generations were studied. In one other family, three affected members of the same generation were studied. The molecular results are shown in fig 1.
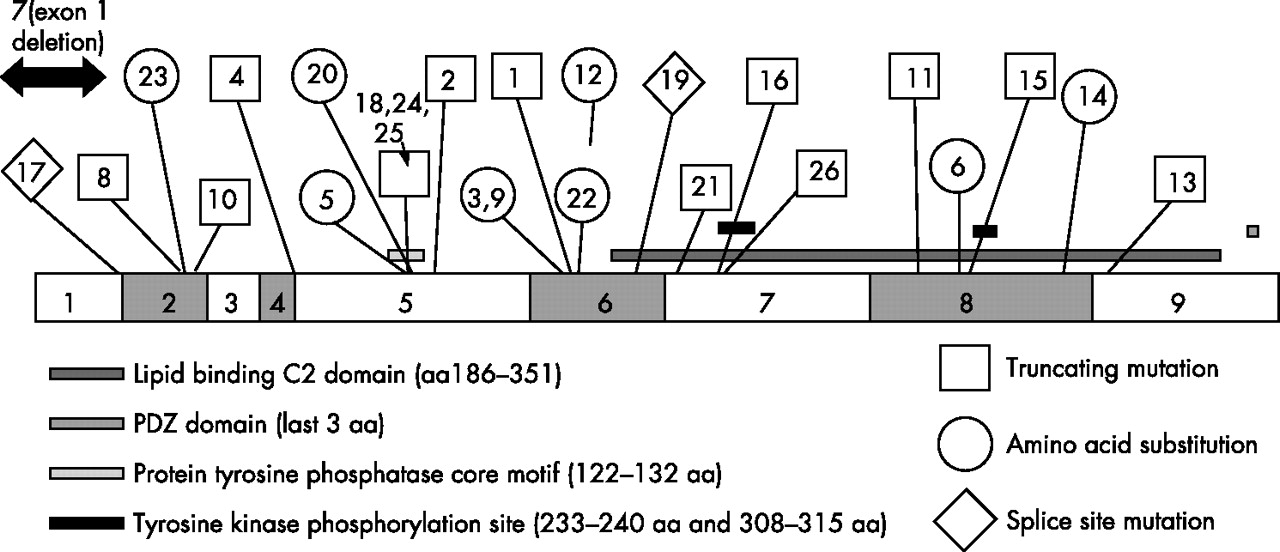
Molecular results. The family reference number is labelled to allow correlation with the tables. There were 22 different mutations within the PTEN gene, in addition to an entire exon 1 deletion in a patient who met the CS criteria. Mutations were not clustered in any one part of the gene. Three mutations were in the PTPase core motif in exon 5, but no point mutations were identified in the first or third exons. There are 15 previously unreported mutations, including one in exon 9. Where proven, 9 of the mutations were de novo, 7 mutations were paternally inherited and 3 were maternally inherited.
Clinical findings
Presenting clinical features are described in fig 2.

{kind=link}
{kind=link}
Presenting clinical features of probands. All childhood probands presented with motor delay, macrocephaly and learning difficulties. The first bar represents those children who presented with minimal signs of macrocephaly and motor delay/learning difficulties. Two male patients included in this bar had penile freckling noted at diagnosis, but had no lipomata or hamangiomata. For one participant, seen as an adult, the major diagnostic clue was his presentation aged 15 with a thyroid follicular cancer. Only two adults had learning problems and a history of early motor delay; one with Lhermitte–Duclos disease and the other with haemangiomas and lipomas. Three adults presented to a genetics service following a tumour diagnosis: one with a bowel adenocarcinoma in situ in a bowel polyp, who had previous severe fibrocystic disease of the breasts, necessitating bilateral mastectomy; one with a breast cancer, with a history of benign thyroid disease; and the third was a woman who presented with a respiratory arrest, secondary to a polyp obstructing her airway. The diagnosis in the third case was made on the basis of dermatological signs, and she subsequently developed breast cancer and benign thyroid disease. Only one adult (in whom the diagnosis was made by a dermatologist) had facial lesions biopsied to confirm the presence of trichilemmomas, which demonstrates the difficulty of using the Cowden Consortium guidelines to aid diagnosis in a clinical setting.
Two participants, a father and son (9.2 and 9.1) had Lhermitte–Duclos disease, now considered a pathognomic feature of CS. Each of them met the CS criteria without including this as a pathognomic criterion.
The cutaneous signs seen were acral keratoses, palmar and plantar keratoses and facial papules, which were confirmed as trichilemmomas in one patient (7.1). We have listed where >6 lesions were seen, as this is the number required by the Cowden Consortium criterion for a diagnosis.
All but three adults who met the CS criteria also met the Parisi28 BRRS criteria (tables 2, 3). The eldest non-proband, aged 75 years, had bowel polyps, facial trichilemmomas, skin tags and three pitted keratoses on the plantar surface of the left foot, but did not have sufficient mucocutaneous signs to meet the CS criteria. The oldest of the four participants with no mucocutaneous signs of CS was aged 8 years and 7 months. Adult participants reported that the mucocutaneous features had increased in quantity and severity with age.
Analysis of mean and median ages at which participants met criteria for Cowden syndrome
We specifically enquired about delay in walking (>18 months of age). Fifteen individuals (35.7%) were reported to not have walked before 18 months of age. Four non-probands (24%) had delayed walking. Only two individuals who reported walking late did not subsequently need extra support in school. These data are based on recall and so should be interpreted with caution.
Macrocephaly
All (100%) participants had a head circumference >99th centile for age, including all non-probands. Two adult probands reported that they were born following a difficult labour, attributed to large head size. Five participants had macrocephaly noted on antenatal scan and in two of these cases, an elective caesarean section had been planned as a result. Nine participants, including six adults, had investigations as infants to understand the cause of their macrocephaly. Seven children had cranial MRI performed: three reports comment on large or ”generous” ventricles, but none had hydrocephalus, and the other four had normal MRI scans.
Tumours
Malignancies observed in the series are reported in table 4. The average age of women without breast cancer (excluding 1.1, who had undergone bilateral mastectomy for fibrocystic disease) was 40.9 years. The breast lump in 4.1 was found 1 month after mammography screening. The schwannoma in 1.1 was on the left tibial nerve and presented with debilitating pain. It was detected on an MRI scan and surgically removed, after which the pain subsided.
Analysis of percentage of participants that meet Cowden Syndrome (CS) criteria by age
Malignancies observed in this series
DISCUSSION
This cross-sectional clinical study provides evidence that the earliest features of the phenotype associated with PTEN mutations are macrocephaly and hamartomas, with the mucocutaneous features and sometimes malignancies developing over time in the same patients. It brings into question whether the division into BRRS and CS is of clinical utility.
Absence of a genotype–phenotype correlation
Patient 7.1, who met the CS criteria, had a deletion of exon 1 detected by multiplex ligation-dependent probe amplification testing, thus providing further evidence to refute a genotype–phenotype correlation. Whole exon deletions have previously only been reported in BRRS, but not in CS.39,44 This patient provides evidence that it is appropriate to look for exon deletions in patients with a diagnosis of CS adding further weight to our assertion that it is unhelpful to split PTEN-related disorders into separate clinical syndromes.
In this study, point mutations were identified throughout the gene except in exons 1 and 3. Features differed from person to person within families. For example, in family 11, the proband presented with macrocephaly, penile freckling and speech delay, but his mother had mucocutaneous features of CS with benign thyroid disease, and also had BRRS features, including haemangiomata and lipoma. There was a family history of thyroid disease, Lhermitte–Duclos disease and breast cancer. This family could be described as an “overlap” family, but we suggest that such families are simply showing most clearly the variable expression and age-related penetrance. Furthermore, variable phenotypes were seen in patients from different families but with the same mutation—for example, p.Ser170Arg was seen in families 3 and 9 (table 1).
It is worth highlighting a number of previously reported mutations, seen in this series, to further point out the lack of genotype–phenotype correlation. There were five people with the p.Arg130X mutation (three probands and two of their relatives), all of whom have features of BRRS and in whom there was a progression of skin findings with age. Three children <5 years of age had no mucocutaneous features of CS, one 13-year-old had <6 typical mucocutaneous features, but a man aged 37 years met the diagnostic criteria for CS, providing a pertinent example that the two conditions are on a continuum. Other cases with this mutation are reported.2,28,45,50 The mother and son described by Zori et al were reported as having CS and BRRS respectively; however, at the age of 18 years, the son developed a thyroid cancer, typical of CS. One family described in detail by Parisi et al had BRRS (family 1); a father and four of his five children were affected, but no skin findings are documented in the father.10,28 Another example is the p.Asn48Lys mutation; in our study, this was seen in a boy (23.1) with learning difficulties and lipomata, which was previously reported in a patient with many features of CS.51
It has recently been reported that splice variants may have different downstream effects that influence phenotypes.43 In this series, there were two splice-site mutations, both de novo (patients 17.1. and 19.1). Both patients have a severe phenotype for their age, including features of CS as children (table 1). The c.634+5G→A mutation was reported previously in two maternal half-brothers with BRRS. They were young (3 years and 7 months and 5 years and 2 months)at the time the paper was written.28 The c.164+1G→A mutation has been previously reported in a family with CS.52 There were too few patients with splice-site mutations in our series for us to address this possibility further.
Diagnostic criteria for BRRS and CS lead to sex-biased diagnosis
Using Marsh’s criteria, 46% of male participants in this study could be classified as having BRRS, whereas only 6% of female participants fulfilled this strict definition. Neither of the two female childhood probands met Marsh’s BRRS criteria. Penile freckling is a major feature, and so we believe that these results reflect the bias of the diagnostic criteria towards diagnosis in males rather than a true higher incidence in males. Use of Parisi’s criteria lead to less sex-biased diagnosis, with 85% of males and 75% of females falling within the BRRS category, making these criteria more useful in clinical practice when deciding which family members to test. It is difficult to formulate criteria to recognise all people with a PTEN mutation, especially in young children; 78% of children in this study would be identified using the Parisi definition. Three probands were tested on the basis of macrocephaly and early motor delay alone in the absence of other findings, showing that clinical judgement is required in the face of minimal features, especially in younger female patients.
CS is reported to have a female preponderance, and in contrast to BRRS the criteria include diseases more common in or affecting only women (breast and endometrial cancer). In this study, 75% of female participants met the diagnostic criteria for CS, compared with only 42% of male participants. Although this may reflect the young age of many males in the study, the reporting in the literature of more female patients with CS may reflect ascertainment bias inherent in the diagnostic criteria. Strict diagnostic criteria are pertinent in research studies, but may result in underdiagnosis in clinical practice.
Age-related penetrance of CS/BRRS features
Older participants were more likely to meet the CS criteria, reflecting age-related penetrance of the CS features. Children who met the CS criteria had multiple hamartoma formation, rather than florid mucocutaneous features. However, many children had some mucocutaneous features of CS, but not in sufficient quantity to make a diagnosis based on these features alone. It is reasonable to speculate that increasing age alone is required for patients to fulfil the criteria; many of the features of CS are not apparent until adulthood and therefore, the difference between the two conditions is the age of presentation. In further support of this is our observation that six adult participants had been investigated in infancy or childhood for macrocephaly. If these people were to be investigated today, a diagnosis of BRRS might be considered, adding weight to the notion that BRRS is the earliest presentation of CS, demonstrating the variable expressivity of one condition.
A milder phenotype found in non-probands
Only 70% of adult non-probands met the CS diagnostic criteria (without inclusion of family history). The study of this group showed that a mild CS phenotype can be observed in adulthood, which has perhaps previously gone unrecognised.
Macrocephaly seen in all participants
Macrocephaly was the most consistent clinical feature, seen in all probands, and importantly, in all non-probands also. It is of note that macrocephaly was noted antenatally. Previous studies have suggested that only 38% of patients with CS have macrocephaly or megencephaly,53 although Starink et al, in their study before the discovery of the PTEN gene, observed macrocephaly in 80% of their patients.10 Unfortunately, many reports of patients with CS do not report head circumference. As macrocephaly was observed in all PTEN carriers, head circumference is a useful diagnostic pointer, particularly when deciding which people to test for PTEN on a limited budget. PTEN is a negative regulator of cellular growth and is expressed in the brain. Given that macrocephaly can be present at birth and MRI scanning fails to show any local areas of excessive growth,54 it is likely that this feature is due to a germline heterozygous mutation; it is difficult to hypothesise that a ”second hit” is responsible for this generalised and consistent finding. Macrocephaly is also observed in neurofibromatosis type 1 and cardio-facio-cutaneous syndrome, both autosomal dominant conditions caused by tumour-suppressor genes in the RAS pathway, which interacts with the phosphoinositol 3-kinase pathway.
Differences in incidences of learning problems and motor delay between CS and BRRS are likely to be due to the variable expression of these features
All the childhood probands had been referred for a clinical genetics opinion because of concerns about early motor development or learning difficulties, and this ascertainment bias leads to an overestimate of risk of learning difficulties in children diagnosed with BRRS. This study revealed that 12% (2/17) of non-probands had learning difficulties, and although based on small numbers, this figure may be useful to quote for couples contemplating prenatal diagnosis. Although developmental delay is not commonly discussed in CS literature, it is a feature in some patients, including the original report of Cowden.11 In a review of published CS cases, mental retardation was reported in 12% of cases,19 and 15–20% has been suggested in previous studies.28 We believe that the difference in frequency of learning difficulties in patients with BRRS compared with those with CS reflects the fact that children with macrocephaly and no learning or motor problems are unlikely to come to medical attention as children, and that this difference simply demonstrates variable presentation, a well-recognised phenomenon in other autosomal dominant conditions.
Tumour and malignancy incidence
Few conclusions regarding tumour incidence in PTEN-related disorders could be made from this study, owing to small numbers. Of the adult non-probands in this study, 2/14(14%) had cancer. The eldest non-proband was free of malignancy at the age of 75 years.
In this series, 15% of adult female probands, but no non-probands had breast cancer. One case of breast cancer was a rare squamous cell cancer, which developed between screening episodes.
In our study, the presence of non-malignant thyroid disease, in the form of thyroid nodules, was high even in non-probands (60%). This was mostly detected on screening. It is of interest that in family 8 (c.141delG), in contrast to any other family in the study, there was a strong family history of malignancy and that the only two thyroid cancers occurred in this same family. Freeman et al55 identified that the onset of tumour formation in PTEN-deficient mice was influenced more by genetic background or modifier genes and less on the type of mutation. This needs further investigation in humans.
One patient had a pre-malignant CIN 3, cervical intraepithelial neoplasia, stage 3 of the cervix detected on routine screening. Cervical cancer is not a known component of CS or BRRS, but loss of heterozygosity and intragenic somatic mutations have been demonstrated in primary cervical cancer tissue.56 One patient developed a granular cell tumour (also known as a granular cell schwannoma). Neural origin tumours have been reported in association with PTEN mutations.10,37
Screening
Previous studies have suggested that children with BRRS should be monitored for the malignant components of CS as they approach adulthood. Although our study does not contribute to the evidence basis for such a statement, the evidence that these two conditions are variable manifestations of the same condition suggests that surveillance should at least be discussed and considered at this stage.
CONCLUSION
Most BRRS cases are reported in children by paediatricians and most CS cases in adults by dermatologists and oncologists. Current literature still tends to differentiate between CS and BRRS and seeks a genotype–phenotype correlation. This is despite the fact that both conditions can be caused by the same mutation in the same gene, can be present in different members of the same family (so called overlap families), and have many clinical features in common. It is only with the advent of a diagnostic test that the true incidence and breadth of phenotype of PTEN-related disorders is being recognised. Interpretation of the data in this study suggests that the division into two separate conditions is based on historical reporting and is no longer relevant. Careful clinical phenotyping in this study leads us to suggest that the features represent age-related penetrance of the same condition, which is a well-recognised phenomenon in other autosomal dominant tumour-suppressor disorders such as neurofibromatosis type 1. Removing the distinction between CS and BRRS has practical as well as intellectual merits. In providing further evidence that these conditions are artificially divided, support is gained for the notion that screening programmes appropriate to one subset should be adopted by the whole group.
Many cases in this study would not have been identified if clinicians had adhered to the existing strict diagnostic criteria. In our study, we found macrocephaly to be a consistent finding (100%), as are mucocutaneous features (90%), and both are helpful when deciding which people to test. Children, particularly girls with motor delay or learning difficulties with macrocephaly, should be considered for testing. Predicting prognosis and tailoring screening regimens for people known to have a PTEN mutation remains problematic and underlines the need for a rigorous long-term follow-up study, which is currently under way.
Acknowledgments
This work was supported by the Birth Defects Foundation (Newlife). We thank all the families who took part in the study and the clinicians who have supported the study. We also thank A Hallsall, K Becker, J Berg, C Brewer, J Bruce, P Carroll, T Cole, W Doak, D Eccles, S Ennis, G Evans, R Fisher, S Gallacher, A Golash, J Greene, S Hodgson, L Izatt, B Kerr, C King, H Kingston, M Knight, F Lalloo, S Leonard, S Mansour, B Mellors, V Murday, R Newbury-Ecob, A Norman, R Park, A Parker, M Patton, N Rahman, L Raymond, E Reid, L Side, H Stewart, J Tolmie, and L Wilson.
REFERENCES
Footnotes
-
Published Online First 25 May 2007
-
Competing interests: None declared.