Article Text

Abstract
Background: CHARGE syndrome is a non-random clustering of congenital anomalies including coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies, and deafness. A consistent feature in CHARGE syndrome is semicircular canal hypoplasia resulting in vestibular areflexia. Other commonly associated congenital anomalies are facial nerve palsy, cleft lip/palate, and tracheo-oesophageal fistula. Specific behavioural problems, including autistic-like behaviour, have been described. The CHD7 gene on chromosome 8q12.1 was recently discovered as a major gene involved in the aetiology of this syndrome.
Methods: The coding regions of CHD7 were screened for mutations in 107 index patients with clinical features suggestive of CHARGE syndrome. Clinical data of the mutation positive patients were sampled to study the phenotypic spectrum of mutations in the CHD7 gene.
Results: Mutations were identified in 69 patients. Here we describe the clinical features of 47 of these patients, including two sib pairs. Most mutations were unique and were scattered throughout the gene. All patients but one fulfilled the current diagnostic criteria for CHARGE syndrome. No genotype-phenotype correlations were apparent in this cohort, which is best demonstrated by the differences in clinical presentation in sib pairs with identical mutations. Somatic mosaicism was detected in the unaffected mother of a sib pair, supporting the existence of germline mosaicism.
Conclusions:CHD7 mutations account for the majority of the cases with CHARGE syndrome, with a broad clinical variability and without an obvious genotype-phenotype correlation. In one case evidence for germline mosaicism was provided.
- CHARGE syndrome
- CHD7
- clinical spectrum
Statistics from Altmetric.com
CHARGE syndrome (OMIM 214800) is a pleiotropic disorder comprising of coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies, and deafness. A consistent feature in CHARGE syndrome is semicircular canal hypoplasia resulting in vestibular areflexia.1–3 Other commonly associated congenital anomalies are facial nerve palsy, cleft lip/palate, and tracheo-oesophageal fistula. Specific behavioural problems, including autistic-like behaviour, have been described.4,5 The combination of abnormalities initially known as CHARGE association was first reported independently by both Hall and Hittner et al in 1979,6,7 after which Pagon and colleagues proposed the acronym CHARGE in 1981.8 CHARGE syndrome is an autosomal dominant syndrome with an estimated prevalence at birth between 1 per 10 000 and 1 per 15 000.9 Recent epidemiological data revealed the occurrence of CHARGE syndrome in 1 in 8500 live births in the Atlantic Provinces of Canada.10
CHARGE syndrome is a phenotypically heterogeneous syndrome clinically diagnosed using criteria which have been refined several times. Blake et al suggested diagnostic criteria in 1998.9 A refinement of these criteria for different age groups was proposed to capture the continuum of the presentation of CHARGE syndrome.10 Simultaneously, Verloes suggested an update of diagnostic criteria, emphasising the most specific embryological defects while avoiding non-specific or secondary anomalies.11 He also suggested the exclusion of sex dependent criteria. Both sets of diagnostic criteria are given in table 1.
Updated diagnostic criteria for CHARGE syndrome
CHARGE syndrome was only recently reconsidered to be a syndrome instead of an association after our group discovered CHD7 on chromosome 8 (8q12.1) as a major gene involved in this syndrome.12CHD7 encodes a protein of the chromodomain (chromatin organisation modifier) family. Members of this family share a unique combination of functional domains consisting of two N-terminal chromodomains, followed by a SWI2/SNF2-like ATPase/helicase domain and a DNA binding domain.13,14 It is assumed that CHD protein complexes affect chromatin structure and gene expression and, thereby, play an important role in regulating embryonic development.
We report a study of the phenotypic spectrum in 47 patients with a CHD7 mutation, with special emphasis on differences in presentation in sib pairs that share identical mutations.
METHODS
Patients
The coding regions of the CHD7 gene were screened for mutations in 107 index patients with clinical features suggestive of CHARGE syndrome. In 69 of these patients a mutation was identified (65%), and for 47 patients (22 males, 25 females, two sib pairs) sufficient clinical data were available to include them in further studies. The cohort includes 15 patients reported in our previous study.12 Parental DNA samples of 22 patients, including one sib pair, were tested for de novo occurrence.
Clinical information concerning the patients was obtained through investigation in our own department or through a written questionnaire submitted when DNA of the patient was referred to the DNA diagnostics section of our department. Additional information was obtained from clinicians when necessary. The diagnostic criteria of Blake and Verloes (table 1) were applied to all cases for which sufficient clinical information was available.9,11
All patients or their legal representatives gave informed consent for the DNA studies and the collection of clinical data.
Mutation screening
DNA was isolated according to standard procedures. The 37 coding exons of the CHD7 gene (exons 2–38, accession number NM_017780) and their flanking intron sequences were amplified by polymerase chain reaction (PCR). Subsequently, sequence analysis was performed using a 3730 automated sequencer (Applied Biosystems, Foster City, CA).
The primer sets used previously were optimised by using shorter PCR products to exclude allele dropout.12 Primer information and PCR conditions are given in supplemental tables I and II, available at http://www.jmedgenet.com/supplemental.
Whole gene deletions were excluded by multiplex ligation dependent probe amplification (MLPA). Specific probe sets were designed for exons 2–11 and exons 33–38. MLPA analysis was performed according to the instructions of the manufacturer (MRC-Holland, Amsterdam, the Netherlands; www.mlpa.com). Probe information is given in supplemental table 3 available at http://www.jmedgenet.com/supplemental.
RESULTS
CHD7 mutation analysis
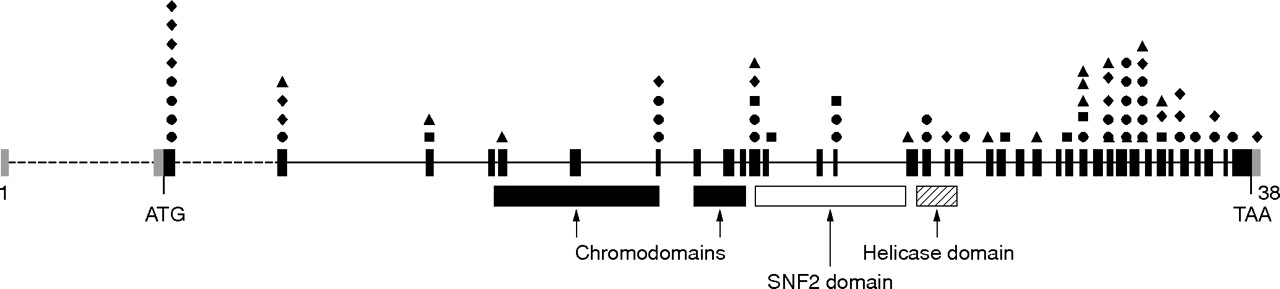
Mutation analysis in our series of 107 index patients revealed 69 mutations in the CHD7 gene (fig 1 and table 2). Two mutations were recurrent, and all others were unique. We detected 31 nonsense, 17 frame shift, 13 splice site, and 8 missense mutations scattered throughout the gene. In the affected sibs identical mutations were identified. A female CHD7 positive patient (no. 22) had a previously identified chromosome 22q11 deletion. Fifteen patients were reported in a previous study,12 and in six of them the mutation was not detected initially. However, after a more thorough investigation with improved primer sets, CHD7 mutations were detected in these patients. The parents were studied for 21 index patients. In 20 cases, the mutation was proven to be de novo. In the sib pair consisting of two boys, mosaicism for the CHD7 mutation was identified in the mother. In the remaining 38 mutation negative patients, whole gene deletions were excluded by MLPA analysis.
Overview of CHD7 mutations

Distribution of CHD7 mutations identified in the 69 CHARGE syndrome patients. Coding exons are indicated in black bars, whereas the non-coding sequences are indicated in grey. Mutations are schematically shown above the exons in which they are located. Nonsense mutations are represented by • (n = 31), missense mutations by ▪ (n = 8), frameshift mutations by ⧫ (n = 17), and splice site mutations by ▴ (n = 13), respectively.
Clinical features
Information obtained through our own investigation and/or through written questionnaires, supplemented with additional information from clinicians, resulted in a description of the clinical features of the 47 selected patients as outlined in tables 3 and 4. Details of these features are provided below. All 47 cases were included in the evaluation unless stated otherwise. The diagnostic criteria of Blake and Verloes (table 1) could be applied to 38 cases.9,11 There was only one patient who did not fulfil either set of diagnostic criteria.
Anomalies in 47 CHD7 positive patients
Further anomalies in 47 CHD7 positive patients
Sufficient clinical information could also be obtained for 23 of the 38 CHD7 negative patients; only two of these patients fulfilled the clinical diagnostic criteria of Blake and Verloes.9,11
A summary of all clinical data of the 47 CHD7 positive patients is given below. A detailed case report is then provided of a girl who did not fulfil the diagnostic criteria of Blake and Verloes (table 1)9,11; the intra-familial variability in sib pairs is also delineated.
Neonatal period
The median gestational age of the patients was 38.2 weeks (n = 45, range 30–42 weeks). Only one patient was reported to be small for gestational age, while feeding difficulties were reported in 33 (70%) patients. Four patients required a gastrostomy due to severe feeding problems.
Four patients died during the neonatal period, three during the first half year of life, and one at the age of 14 years. At the time of investigation four patients were below the age of 1 year.
Coloboma of the eye
A coloboma of one (n = 4) or both (n = 29) eyes was present in 33 patients (70%). As the iris was involved in only nine patients, the coloboma was only visible by fundoscopy in most patients. In none of the patients was the coloboma restricted to the iris only. Microphthalmia was present in ten patients (21%).
Congenital heart defects
Thirty one (66%) patients had a congenital heart defect. Fourteen (30%) patients had major heart defects: six tetralogy of Fallot, two double-outlet right ventricle (one combined with hypoplastic left heart and AVSD), three isolated hypoplastic left heart syndrome, one hypoplastic right heart syndrome, one agenesis of the pulmonary valve combined with hypoplastic left heart, and one Shone’s complex. A right descending aorta was present in three patients and one patient had a vascular ring. The other patients had solitary patent ductus arteriosus beyond infancy (n = 3), patent ductus arteriosus combined with atrium septum defect, and/or ventricular septum defect (n = 6) or a solitary septal defect (n = 4).
Retardation of growth and development
A height below the third percentile was reported in 21 out of 32 patients (63%).
Speech development varied from mild speech delay to severe retardation without speech. Learning disabilities were reported in 24 (75%) out of 32 patients who were above the age of 12 months at last examination. Eight patients (25%) had no cognitive impairment.
Endocrine and urogenital abnormalities
At the time of CHD7 testing, 15 patients (eight girls, seven boys) were over 15 years of age. Gonadotrophin deficiency was present in seven (88%) of these girls, and six (86%) of these boys. Two girls had their menarche at age 14. A hypoplastic uterus was found by ultrasound investigation in three girls. Of all 22 mutation positive boys, four (18%) had cryptorchidism, six (27%) had micropenis, and seven (32%) had both cryptorchidism and micropenis.
Three patients had a horseshoe kidney and in two patients agenesis of the left kidney was demonstrated. A vesicoureteral reflux was reported in three patients and one patient had renal cysts.
Ear and vestibular abnormalities
Dysmorphisms of the ears were noted in all patients, ranging from typical CHARGE ears (small, square, low set, and protruding) to minor structural abnormalities such as absence of an earlobe. One patient had a pre-auricular pit and one patient had narrow external auditory canals. Hearing impairment was demonstrated in 37 out of 41 patients (90%). In 27 patients severe bilateral hearing impairment was observed, while five patients showed asymmetric hearing impairment with unilateral normal or mild hearing loss.
In all 21 patients who underwent CT scanning of the temporal bones, agenesis of the semicircular canals was demonstrated. Vestibular areflexia was demonstrated in two more patients and four patients had a history of balance disturbances. In total, therefore, 27 patients (57%) had some evidence of vestibular anomaly. However, information on this subject was not available for the remaining patients, although motor delay (possibly due to vestibular areflexia) was present in all cases on direct questioning.
Nasopharyngeal abnormalities and clefting
Choanal atresia was present in 17 patients (36%) and was unilateral in only three of them.
Respiratory insufficiency during the neonatal period was reported in 24 patients (51%), 22 of whom had either choanal atresia or a congenital heart defect or both. Tracheomalacia was present in one patient.
Clefting was present in 17 patients (36%): 11 had a cleft lip and palate, five had an isolated cleft palate, and one had an isolated cleft lip.
Gastrointestinal abnormalities
Eight patients (17%) had oesophageal atresia, which in three was accompanied by a tracheo-oesophageal fistula. Two patients had a diaphragmatic hernia and one anal stenosis.
Neurological abnormalities
A minority of patients (n = 4, 9%) had central nervous system abnormalities, including corpus callosum agenesis combined with cerebellar hypoplasia (n = 1), hydrocephaly (n = 2), and atrophy of the cerebral cortex (n = 1). Five patients had convulsions.
Facial nerve palsy was present in 10 patients (21%) and mostly (nine out of 10) involved the right-sided facial nerve.
Skeletal abnormalities
Scoliosis was demonstrated in six patients (13%), kyphosis in one, and abnormalities of the vertebral bodies in three (6%). In one patient a triphalangeal thumb was demonstrated.
Aspecific CHARGE syndrome
Patient 28 (born at 40 weeks’ gestation; birth weight 3700 g, 70th centile, fig 2) was a 5 year old girl with developmental delay, slightly dysmorphic ears, and severe hearing impairment. CT scan showed bilateral agenesis of the semicircular canals. She required a gastrostomy due to severe feeding problems and had surgery on a congenital vascular ring. Her height was at the third centile. No choanal atresia, cleft palate, or coloboma could be detected. In this girl, the only individual in our CHD7 positive series who did not fulfil the current diagnostic criteria for CHARGE syndrome (table 1),9,11 a de novo nonsense mutation was identified, 5833C>T (R1945X) in exon 29 of CHD7.

Patient 28, who is CHD7 mutation positive but does not fulfil the diagnostic criteria (see text). (Written consent was obtained for the publication of this picture.)
Familial cases
Two sib pairs were included from two families. In both cases, identical CHD7 mutations were identified in the two sibs. Interestingly, in both cases the affected sib pairs showed distinct clinical features.
Sib pair 1 were monozygotic twin sisters born at 35 weeks’ gestation (patients 26 and 27; tables 3 and 4, fig 3A,B). Patient 27, who had a birth weight of 1500 g (5th–10th centile), died 29 h after birth due to a combination of hypoplastic left heart syndrome and bilateral choanal atresia. She also had a tracheo-oesophageal fistula and typical CHARGE ears. A hearing test was not performed. There were no colobomata of the irides.
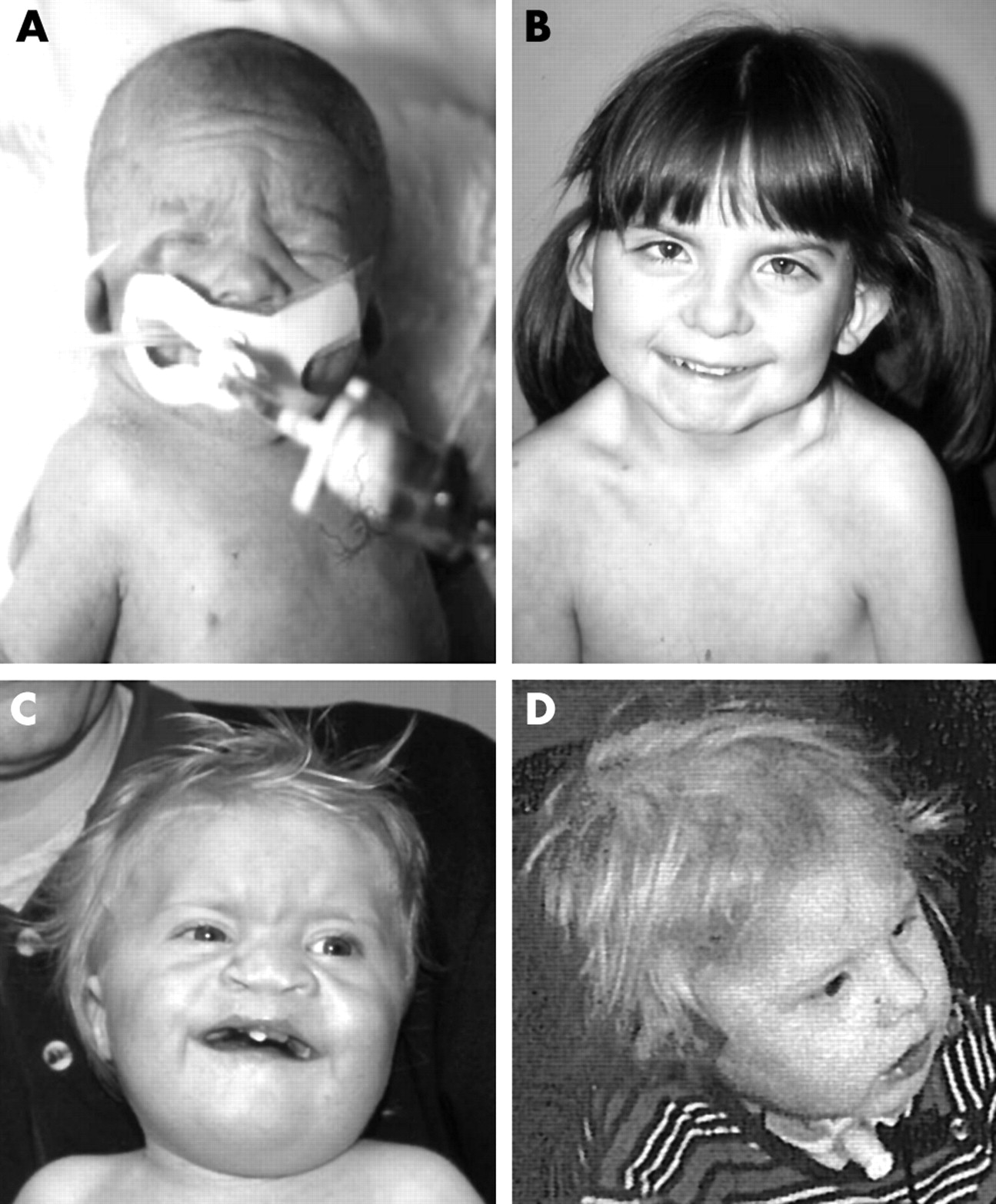
(A) Twin 1 of sib pair 1 (patient 27 in tables 3 and 4), who died shortly after birth; (B) twin 2 of sib pair 1 (patient 26 in tables 3 and 4) at the age of 7 years; (C) sib 1 and (D) sib 2 of sib pair 2, both at the age of 2 years (patients 29 and 30 in tables 3 and 4). (Written consent was obtained for publication of these photographs.)
Patient 26 had a birth weight of 1910 g (25th centile). When examined at the age of 12 years, she had short stature (<3rd centile) and was functioning 4 years behind her chronological age. She was born with a large patent ductus arteriosis that required surgery and she needed numerous procedures to correct bilateral choanal atresia. Her first years of life were complicated by feeding problems, for which she had a gastrostomy until the age of 6 years. She had severe bilateral deafness, abnormal external ears like her twin sister, and bilateral chorioretinal colobomata with a right-sided iris coloboma and an unusual inferior pigment pattern in her left iris. Agenesis of the semicircular canals was not tested for by CT scan, but her gait was unsteady.
Zygosity testing with five unlinked markers was performed and the results were consistent with the twins being monozygotic. In both sisters the spectrum of congenital anomalies was caused by an insertion 5752_5753insA in exon 29 of CHD7.
Sib pair 2 consisted of two brothers (patients 29 and 30; tables 3 and 4, fig 3C,D). Patient 29 was 7 years of age (born at 40 weeks’ gestation; birth weight 2867 g, 10th centile). He had surgery for cleft lip/palate and a complex heart defect (DORV, AVSD, hypoplastic left heart). He had short stature (3rd centile) and severe developmental delay. His ears showed the typical CHARGE dysmorphisms and he had bilateral hearing loss and unilateral facial nerve palsy. He had no colobomata or choanal atresia.
Patient 30, who was 4 years younger, had bilateral hearing loss and typical CHARGE ears, a coloboma of the left retina and choroid, and underwent surgery for a tracheo-oesophageal fistula. He had short stature (<3rd centile) and was severely mentally retarded. He had vocal cord palsy. This boy had no heart defect and no choanal atresia.
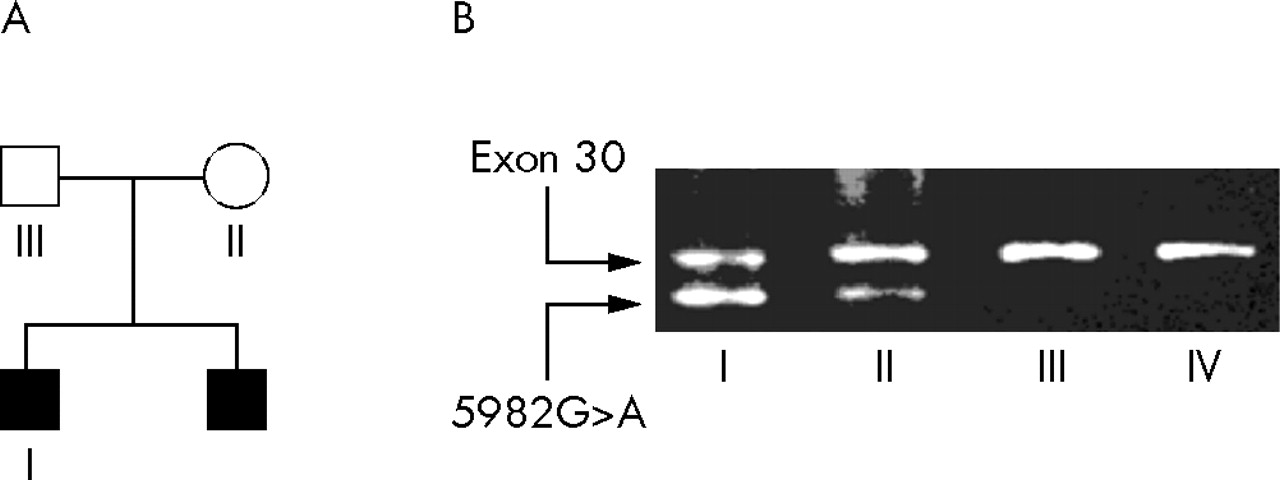
Both brothers have a 5982G>A (W1994X) mutation in exon 30 of CHD7. Sequence analysis of both parents revealed no mutation in the father and a minor aberrant peak in DNA extracted from lymphocytes of the mother. This indicated that a possible mosaicism was present in the mother. This was further investigated and confirmed by an allele specific PCR, using a primer carrying the 5982G>A mutation at the 3′ end, in combination with the regular exon 30 primer set (fig 4). Clinical examination of the mother did not reveal any signs of CHARGE syndrome.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Results of allele specific PCR for the 5982G>A mutation in the family with two affected boys. (B) DNAs of the indicated family members and an unrelated unaffected control (IV) were subjected to a multiplex PCR using a mutation specific primer (5982G>A, lower band) and the regular primer set for exon 30 (upper band). The mutation found in the boys (I) was also present in the mother (II). The different relative amounts of the fragments of individual I and II might reflect the presumed mosaicism in the mother.
DISCUSSION
At the time of evaluation of our clinical data, CHD7 sequencing had been performed in 107 index patients referred to our laboratory because of clinical features suggestive of CHARGE syndrome. Pathogenic mutations were identified in 69 patients (65%), including six patients who had previously tested negative.12 All mutations except two were unique and most mutations had a severe effect on the CHD7 protein, being either nonsense or frameshift mutations (70%).
From both our previous data and a recent report by Arrington et al, it is known that microdeletions of the chromosome 8q12.1 region, including the CHD7 gene, may also result in CHARGE syndrome.12,15 We excluded the presence of such microdeletions in the patients without CHD7 mutations by MLPA. From these results we conclude that whole gene deletions of the CHD7 gene are not a frequent cause of CHARGE syndrome. Currently, we are extending our MLPA analyses in order to assess for the presence of small intragenic deletions.
In 20 out of 21 families a de novo occurrence of the CHD7 mutation could be proven. In the mother of the sibs with the 5982G>A(W1994X) change, this mutation was present as a somatic mosaicism. It is likely that germline mosaicism exists as well. As a consequence, prenatal diagnosis should be offered to all parents of children with an apparently de novo CHD7 mutation.
Of the 69 CHD7 mutation positive patients, 45 index cases were selected for further clinical study together with two sibs, resulting in a cohort of 47 patients. Due to a short follow-up period, clinical information was limited in 11 patients, especially regarding hearing, growth, and development. From the data presented in tables 3 and 4 and the detailed clinical description of our patients, it is clear that within the CHD7 mutation positive subset of CHARGE patients an extensive variability in clinical presentation exists, without any obvious genotype-phenotype correlation. This is best demonstrated in the two sib pairs. In the first sib pair, both twin girls had choanal atresia and a heart defect, but they were discordant for the coloboma and tracheo-oesophageal fistula. The boys of the other sib pair were discordant for cleft lip/palate, heart defect, tracheo-oesophageal fistula, coloboma, and hearing loss.
Missense mutations were found in three patients of the clinical study group, one of whom was mildly mentally retarded. The other two had normal levels of intelligence. However, normal intelligence was also present in five patients with a nonsense mutation. Overall clinical comparison of these three patients with a missense mutation with the rest of the study group did not reveal any clear differences. However, it is still possible that less severe mutations (that is, missense mutations) result in a less specific phenotype, not recognised as CHARGE syndrome. Hence, such patients may not be included in this study. On the other hand, patients with a CHD7 deletion may be more severely affected than patients with a CHD7 mutation, especially if multiple adjacent genes are deleted. Further studies are needed to explore this.
In table 5 the frequency of the main features of CHARGE syndrome in our group of CHD7 mutation positive patients is compared with data from the literature.
The frequencies of characteristic CHARGE findings in a population of CHD7 positive patients compared to the literature
The distribution of features in the clinically diagnosed CHARGE syndrome patients as reviewed by Stromland et al, Issekutz et al, and Tellier et al, is comparable to that in our CHD7 mutation positive patients.3,10,16 This indicates that, within the patient group that fulfils the clinical diagnosis of CHARGE syndrome, there is not a specific subgroup that is more likely to have a CHD7 mutation. None of the clinical features seems to be obligatory for a CHD7 mutation, with the possible exception of vestibular anomalies. Several reports have stressed the high frequency and the high specificity of anomalies of the semicircular canals.1,2,17,18 This was also observed in our cohort of patients. All patients investigated by CT scan or vestibular function tests had either abnormal function or an aplasia of the semicircular canals.
The effect of a CHD7 mutation on a specific organ is variable and does not predict the consequences for other organ systems in which CHD7 is expressed. For instance, a severe heart defect does not exclude normal intelligence (for example, individual 43, tables 3 and 4) and severe mental retardation does not have to be accompanied by severe defects in other organs (for example, individual 8, tables 3 and 4). This results in enormous clinical variability, even within sib pairs.
We carefully tested whether both sets of diagnostic criteria could be applied to our patients (tables 3 and 4).9,11 This was not possible in all cases, since, for example, CT scanning of the temporal bones is required in order to apply the diagnostic criteria proposed by Verloes. For simplification we decided to use the 1998 Blake criteria as listed in table 1 instead of the refined criteria adopted for different age groups.10 Both Blake and Verloes require that at least a coloboma or choanal atresia is present for the diagnosis CHARGE syndrome. Five patients in our study group (individuals 4, 6, 9, 28, and 29 in tables 3 and 4) had neither coloboma nor choanal atresia. Blake et al argued that the choanae are usually patent when orofacial clefting is present and palatal clefting can be substituted for choanal atresia in the scoring criteria.9 As a consequence, only one patient (individual 28 in tables 3 and 4) failed to fulfil the diagnostic criteria for CHARGE syndrome according to both Blake and Verloes. In 38 patients with features suggestive of CHARGE syndrome, no CHD7 mutation and/or deletion was identified. For 27 of these patients, sufficient clinical data were available to apply the clinical diagnostic criteria. Only two of these 27 CHD7 mutation negative patients fulfilled the diagnostic criteria. In both patients aplasia of the semicircular canals was demonstrated. As a consequence, the positive predictive value of the clinical diagnostic criteria is 36/38 (95%). This is substantiated by the fact that after improvement of the sequencing procedure (see supplemental table I available at http://www.jmedgenet.com/supplemental), the mutation positive percentage in our first reported cohort reaches 95% (18/19).12 In the context of the previously suggested genetic heterogeneity,19–21 this is an interesting observation that needs confirmation.
We would like to stress that CHARGE syndrome remains a clinical diagnosis. Although the high percentage of CHD7 mutations in clinically diagnosed CHARGE syndrome patients indicates that CHD7 is the major gene involved, this diagnosis cannot be rejected based on absence of a CHD7 mutation. On the other hand, based on the clinical criteria alone, one CHD7 positive patient would have been missed in our series.
In conclusion, we confirm that mostly unique CHD7 mutations account for the majority of cases with CHARGE syndrome, with a broad clinical variability and without an obvious genotype-phenotype correlation. In addition, we provided evidence for germline mosaicism.
Acknowledgments
We thank the patients and their parents for their participation and D Wieczorek, T Letteboer, J Verheij, M Baars, A van Haeringen, J Cobben, S Maas, W Kok, Y Hilhorst-Hofstee, C de Die-Smulders, M Parisi, V Der Kaloustian, and B Smyle for providing clinical data.
REFERENCES
Supplementary materials
Footnotes
-
Published Online First 14 October 2005
-
Competing interests: none declared
-
Informed consent: informed consent was obtained from all patients whose photographs are reproduced in this article