Article Text

Statistics from Altmetric.com
Recent trials of plasminogen activators in acute ischaemic stroke underscore the delicate balance between promise of benefit and risk. Attempts to manage clinical outcome by systemic infusion of recombinant tissue plasminogen activator (rt-PA) have so far had mixed success.1 2 The benefits seen in the National Institutes of Neurological Diseases and Stroke (NINDS) trial of rt-PA in acute ischaemic stroke were a significant absolute improvement in disability outcome.2 3 However, the risks in that study and in the European Cooperative Acute Stroke Study (ECASS)1 included further disability and mortality associated with haemorrhagic consequences of the plasminogen activator (PA).1 2 Three recent studies of streptokinase in acute ischaemic stroke were terminated because of safety concerns.4-9 In all five trials, the risk of symptomatic haemorrhage associated with the PA was significantly increased over placebo (table 1). In addition, approaches which effect recanalisation of documented cerebral arterial thrombotic occlusions by intravenous or intra-arterial PA infusion have yet to be rigorously shown to promote overall benefit, although anecdotal evidence suggests that individual patients may improve clinically.10-24 The reasons are practical. The perceived risks, those of angiography and the interventional procedures required for intra-arterial PA delivery, have not been fully evaluated,25 although the relative risks of diagnostic angiography are low.26-28 Clearly, this assessment hides many important differences in PA behaviour, study design and conduct, patient populations, diagnostic and therapeutic procedures, disease severity, comorbidity, and a host of other potential contributors. None the less, concerns which may erode the promise of benefit of thrombolytic agents in acute ischaemic stroke include the accentuation of innate risks associated with the evolution of ischaemic injury after the stroke event itself.29-34 These include early mortality caused by severe brain oedema and the development of haemorrhagic transformation which causes clinical deterioration or death.
Odds ratio (OR) analysis of haemorrhagic transformation in trials of thrombolytic agents in acute ischaemic stroke
The nature of these risks and their augmentation by PAs are so far not completely understood. For rt-PAs they seem to encompass the time from onset of ischaemic stroke symptoms to treatment for documented middle cerebral artery (MCA) occlusion,35 diastolic hypertension,36 body mass,36age,37 signs of ischaemic injury at baseline,1 and perhaps other yet to be identified factors. It is clear that a reduction in the frequency of symptomatic intracerebral haemorrhage may substantially improve clinical outcome by reducing one significant contributor to mortality in each of the recent trials.1 2 4-9 29-32 38 The thesis we consider here is that ischaemic injury to the microvasculature, manifest indirectly by early signs of focal ischaemic injury on CT, is central to the risks of oedema and of parenchymal haemorrhage. Here, we posit that appropriate patients with ischaemic stroke will derive further benefit from PAs if the territory and volume of damaged cerebral microvasculature, and thus the risk of oedema formation and symptomatic haemorrhage, is relatively small.
The microvasculature as a target of ischaemic injury
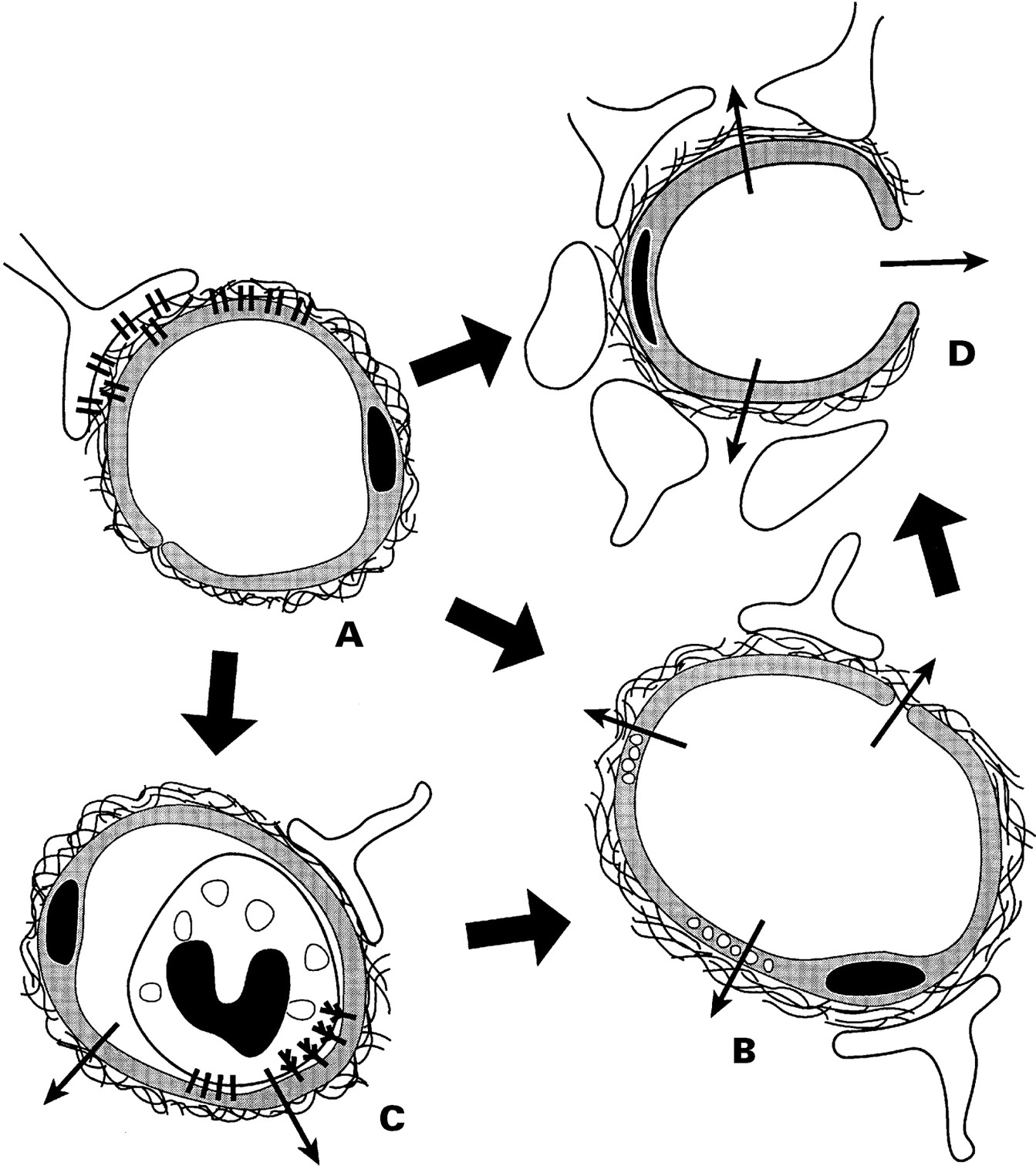
When blood flow through a feeding cerebral artery ceases, the downstream microvasculature is a proximate target. Although non-vascular cells have long been thought to be the primary target of ischaemic insults, events occurring at the blood-vascular-parenchymal interfaces are necessary for the initiation of tissue injury (figure A-D). During experimental focal cerebral ischaemia, with the fall in tissue oxygenation (1) microvascular permeability barriers are lost (figure B)39-42; (2) the microvascular endothelium responds by the sequential expression of leucocyte adhesion receptors (figure C)43-45; (3) the basal lamina and extracellular matrix (ECM) undergo progressive loss of component antigens (figure D)46; and (4) cell-matrix adhesion interactions within microvessels are altered (figure D).47 48

{kind=link}
Schematic diagram of the effect of ischaemia on microvascular permeability and integrity. (A) Normal cerebral microvessel. Endothelial cells and astrocytes bound to basal lamina by integrin adhesion receptors. Blood-brain barrier intact. (B) Breakdown of blood-brain barrier. (C) Leucocyte adhesion by receptors on endothelium and granulocytes; increased permeability caused by granule release. (D) Breakdown of basal lamina with loss of astrocyte and endothelial cell contacts; permeability to large cells—for example, erythrocytes (see text).
Microvascular permeability barriers
The permeability barrier consists of: (1) the blood-brain barrier, represented by the interendothelial cell tight junctions which regulate substrate transfer39 40; (2) the basal lamina, a structural barrier to the extravasation of cellular blood elements49 50; and (3) perivascular astrocytes which make up the parenchymal component of the microvasculature (figure A).51 With the loss of the endothelial cell permeability barrier,39 40 and degradation of basal lamina/ECM components, the brain parenchyma is exposed to the blood plasma and its cellular elements.39 40 46 51-53
The formation of the endothelial blood-brain barrier relies on the interdependence of endothelial cells and astrocytes, as elegantly shown in chick-quail adrenal vascular tissue/brain tissue xenograft and fetal-adult hippocampal/neocortex allograft preparations.54 55 Astrocytes promote microvascular endothelial blood-brain barrier properties including tight junctions, dye exclusion, and antigenic features.40 56 57 Coculture experiments have shown that soluble factors are necessary to maintain some of those barrier characteristics.55 58 59
Intact basal lamina also requires juxtaposition of the microvascular endothelium and astrocytes.60 61 In culture, astrocytes can secrete the basal lamina/ECM components laminin and fibronectin,62 as well as chondroitin sulphate proteoglycan.63 Conversely, collagens stimulate astrocyte induced endothelial cell maturation.64 Microvascular endothelial cell derived ECM components stimulate astrocyte growth and generation of glutamine synthetase.65 66
The integrity of mature cerebral microvessels also seems to involve adhesion receptors which link the cellular components to specific ligands within the basal lamina/ECM. For instance, it has recently been shown that integrin α1β1, a receptor for the ECM ligands laminin-1 and collagen IV, is found on microvascular endothelium,47 and integrin α6β4 is expressed on astrocyte end feet juxtaposed to its matrix ligand laminin-5.48 Hence, development of the blood-brain barrier and the basal lamina depend on the proximity of astrocytes to the endothelium, whereas the integrity of this relation involves cell matrix adhesion molecules.
Focal ischaemia and changes in microvascular integrity
After experimental MCA occlusion, microvascular expression of integrins α1β1 and α6β4 decreases rapidly in the region of focal ischaemia.47 48 A more gradual loss of laminin-1, laminin-5, cellular fibronectin, and collagen IV antigens within the basal lamina/ECM also occurs.46 48 Hamannet al demonstrated a significant correlation between the development of haemorrhagic transformation and the regional loss of basal lamina architecture by 24 hours after MCA occlusion.46 52 67 Hence, haemorrhagic transformation occurs in regions where ischaemia causes a breakdown in the basal lamina, and disruption of adhesion between microvascular cellular components and the internal matrix scaffolding.
Degradation of microvascular barriers
The loss of constituents of the basal lamina and the rapid disappearance of related integrins may reflect down regulation of mRNA or protein synthesis, or degradation of mRNAs or proteins by enzymes secreted by endothelial cells, astrocytes, or circulating cells (figure D). At least three pathways may potentially lead to degradation of components of the basal lamina: (1) activation of plasminogen by endogenous PAs; (2) secretion of metalloproteinases (MMPs); and (3) release of PMN leucocyte specific granule enzymes. Non-ischaemic cerebral tissue generates PA activities.68 More specifically, cerebral microvascular endothelial cells express t-PA in situ69 70 and in vitro,71 whereas astrocytes produce t-PA and u-PA in response to specific mitogens.72Matrix laminins are substrates for plasmin,73 74 and plasmin activates MMP-1 (collagenase) and MMP-3.75 Certain MMPs released from vascular endothelium and leucocytes during the inflammatory phase of cerebral ischaemia employ collagen IV (MMP-1, MMP-2, and MMP-9) and laminin (MMP-2 and MMP-9) as substrates to promote tissue remodelling.76 77 Gelatinases A (MMP-2) and B (MMP-9), which cleave collagen IV, are present in plasma.78 Rosenberg et al have suggested that MMP-9 appears by 24 hours after MCA occlusion in the anaesthetised rat.79 80 Also, enzymes from PMN leucocyte granules, including collagenase (MMP-8), elastase, and cathepsin G, released during their activation, degrade laminins and collagens.81-85 It is postulated that these pathways, unopposed by their inhibitors, may contribute to the dissolution of the basal lamina/ECM and the blood-brain barrier, thereby disrupting the normal relations of endothelium to astrocytes.86 87
Leucocytes and microvessel permeability
Circulating leucocytes may promote changes in microvascular permeability at several levels. Adhesion of PMN leucocytes to specific endothelial cell receptors contributes to loss of microvascular patency,88-91 initiates diapedesis and transit into the parenchyma, and thereby adds to tissue injury (figure C).92 After MCA occlusion, P-selectin, ICAM-1, and E-selectin are expressed on cerebral microvascular endothelium in a distinct sequence.43 44 P-selectin responds to thrombin generated during coagulation activation, histamine, activated complement, and superoxides.93-95 Astrocytes and microglia generate cytokines which stimulate endothelial cell-leucocyte adhesion receptor expression and promote chemotaxis.96-98For instance, ICAM-1 and E-selectin appear in response to interleukin (IL)-1β and tumour necrosis factor (TNF)-α.99 100These events initiate and expand the cellular inflammatory response to ischaemic injury. In addition, IL-1 has been shown to directly mediate neuron injury.96 97 Blockade of IL-1 by exogenous IL-1ra in a rodent MCA occlusion model has been associated with a significant reduction in ischaemic injury at 24 hours.97 During leucocyte transmigration and activation, the respiratory burst generates superoxide free radicals and release of granule proteolytic substances which contribute to blood-brain barrier, basal lamina/ECM, and perivascular tissue degradation.101 Cleavage products of laminins, generated by this process and by plasmins, are potent chemoattractants for leucocytes.102 The presence of complement C5 receptors on astroglial cells103 suggests the possibility that activated complement generated during ischaemic injury may also contribute to leucocyte activation. Free radicals, localised to ischaemic scattered cerebral microvascular endothelial and smooth muscle cells in vivo,104-106 may increase cerebral endothelial permeability and alter adhesion receptor distribution.107 In other systems, tissue haemorrhage accompanies tissue injury with inflammation.108
Barrier loss and haemorrhagic transformation
Extravasation of plasma and blood cells is initiated very early and becomes evident by 24 hours after experimental MCA occlusion in the corpus striatum.52 67 109 Haemorrhagic transformation seems tied to processes which alter the integrity of the blood-brain barrier and the basal lamina. In clinical preparations and certain experimental models, parenchymal haemorrhage has also been attributed to the impact of systemic blood pressure, the downstream movement of thromboemboli, or rupture of larger injured vessels which may have undergone degenerative changes.30 110-113 The distinguishing features are the rapid extravasation of blood leading to the compression of contiguous and adjacent tissues with loss of neurological function.30 110 It is, so far, conjectural whether the processes leading to the development of coagula in the ischaemic zone begin with the microvascular events that lead to haemorrhagic infarction,33 34 52 however, vascular disintegration would seem to be necessary.
Several well known conditions which are aggravated by advancing age, long term hypertension, and diabetes target the microvasculature and may contribute to loss of microvascular integrity.114These include lipohyalinosis or microangiopathy, which has been classified in three states of damage severity:115 (1) sclerotic and hyalinotic thickening of the vessel wall with onion skinning, which primarily affect arterioles 100–200 μm diameter116; (2) additional disorganisation of the vessel wall and disruption of the internal elastic lamina with occasional foam cells (these changes may be associated with haematoma formation)117; and (3) additional fibrinoid degeneration of the vessel wall with thrombosis.118 Importantly, deposition of amyloid in the vessel wall (amyloid angiopathy) leads to loss of vascular wall integrity, leakage, and later haemorrhagic changes.119-123 Most experimental models of focal ischaemia do not mimic these age-dependent vascular disturbances, which may also explain the rare occurrence of large haemorrhages in animal models compared with humans. Hence, loss of microvascular integrity may connect the experimental findings of blood cell extravasation to clinically important haemorrhagic transformation.35
Oedema formation
During focal cerebral ischaemia, increased microvascular permeability also leads to the entrance of fluid and plasma proteins into the injured region (figure B).124 In selected models, exposure of coagulation factors to perivascular tissue factor (TF) results in intravascular and extravascular fibrin deposition.125 126 Here, thrombin is generated which may increase endothelial cell permeability.127-129 Platelet activating factor (PAF), TNF-α, and bradykinin released during focal ischaemia also increase endothelial permeability.130-132Furthermore, tissue haemorrhage itself may promote changes in permeability of the blood-brain barrier and oedema formation.133
Brain oedema develops immediately when regional cerebral blood (rCBF) falls below the threshold of structural integrity at 10–15 ml/100 g/min.124 134-136 In a feline model, within 4 hours of MCA occlusion, cortical water content increased steadily from 80.7% to 83.0% wet weight.134 This 2.3% net uptake of water was accompanied by increased tissue Na+ and decreased tissue K+ concentrations, a shift of water from the extracellular into the intracellular compartment, and a linear increase in brain volume.134 Fluid entry involves osmotic and ionic gradients between blood and ischaemic brain tissue, and pinocytosis of water in the presence of residual plasma flow.137-140Swelling within the ischaemic zone is explained by the failure of energy dependent membrane ion exchange pumps, subsequent influx of Na+ and water into the intracellular compartment, resulting in cell hydrops at the expense of the size of the extracellular space.141 In the early stages of experimental focal ischaemic injury, increased transport of plasma markers is associated with increased pinocytosis in brain vascular endothelial cells consistent with attempts by the vasculature to compensate for decreased substrate delivery.142 143 Here, the ultimate source of the expanded intracellular water compartment is vascular.
In focal ischaemia, the accumulation of tissue water may not resolve if reperfusion is achieved later than one hour after arterial occlusion.137 Moreover, reperfusion can further enhance oedema in areas of dense ischaemia.137 Subsequent studies in focal cerebral ischaemia indicate that PMN leucocytes may contribute further to tissue water accumulation in the injured territory.144-146 Taken from studies of oedema in the skin,147 it is possible that alterations of adhesive processes that bind neurons to their glial neighbours may further promote oedema formation.
Oedema formation and early radiological signs of ischaemia
The net uptake of water by the ischaemic brain tissue affectsx ray attenuation.148 149 A 1% increase in content of tissue water causes a decrease ofx ray attenuation by 2 to 3 Hounsfield units (HU).150 After experimental MCA occlusion, tissue water uptake is detectable by CT which shows a linear decline inx ray attenuation by about 1.5 HU/h.151 Because of the normal noise in each CT image, a change of about 5 HU is necessary for an increase in tissue water to be visible.
Shrinkage of the extracellular space causes a decrease in proton diffusion which is best detected by diffusion weighted MRI.150 152 The detection of ischaemic changes by diffusion weighted MRI is immediate.153 The hyperintense area on the MRI representing diminished proton diffusion may be reversible during the first 2 hours after arterial occlusion, but later may correspond to irreversible tissue damage.154Therefore, the presence of visible subtle hypoattenuation on CT is consistent with a minimum 2% focal uptake of water and an arterial occlusion which occurred some time earlier. The degree of hypoattenuation may be confounded by an increase in local cerebral blood volume, a well known compensatory mechanism for low perfusion pressure.155
From clinical experience, cerebral tissue hypoattenuation is regularly visible within the first 6 hours after the onset of ischaemic symptoms. The loss of the permeability barriers and entry of intravascular fluid, together with nonvascular cell injury contribute to early signs of ischaemic injury notable by CT: a regional decrease inx ray attenuation and evidence of structural changes, marked by mass effect. This hypoattenuation can at an early stage delineate a volume of parenchymal tissue that will subsequently become necrotic and, if exceeding 50% of the MCA territory, is associated with a mortality of 85%.156 157 It seems likely, but remains unestablished, that even very subtle hypoattenuation on CT and areas with restricted diffusion detected by MRI indicate oedema and irreversible tissue damage caused by sustained focal hypoperfusion. Whether oedema formation and microvascular haemorrhage are two manifestations of the same microvascular injury processes is of central relevance.
Vascular contributors to ischaemic damage
The degree of ischaemic injury in a cerebral arterial territory also depends on the intrinsic vulnerability of the tissue within that region. The volume of ischaemic cerebral tissue may depend on (1) protection by the collateral circulation (regional vascular flow characteristics), and (2) the vulnerability of selected non-vascular cells (selective cellular vulnerability) to the vascular territory at risk.
Experimental studies and clinical experience suggest that the corpus striatum is more sensitive to ischaemic injury than the overlying cortex following proximal MCA occlusion.158-160 This differential sensitivity is partly explained by vascular anatomical conditions: The cortex is protected by a net of leptomeningeal arteries which communicate with the parenchymal arterioles and between cortical territories, whereas the circle of Willis offers a continuous shunt of the basal brain supplying arteries.161 Furthermore, the microvascular network of the cortex is distinctly different from that of the corpus striatum. Developmental features of the rat cortical microvasculature, described by Bär et al, indicate a roughly three dimensional hexagonal network extending from the pial vessels to the grey matter-white matter border.162 163 This arrangement provides regional collateral vascular circuitry to shunt blood locally.164In the non-human primate and in humans, flow is directed from the MCA to the corpus striatum through lenticulostriatal arterial (LSA) perforators. Here, erythrocyte transit time is increased,165 so that the regional CBF is closer to the threshold for ischaemic injury. In human patients and primates, the corpus striatum is particularly subject to unrecoverable injury after obstruction of the proximal (M1) MCA.159 Occlusion of the proximal MCA causes an immediate drop in blood flow in the territories supplied by the LSA, whereas the superficial cortex may be salvaged by leptomeningeal collaterals.166 In non-human primates after proximal MCA occlusion, haemorrhagic transformation is most often seen in the corpus striatum, where ischaemic injury is greatest.109
Selective vulnerability of neurons and glial cells is readily evident during experimental global cerebral ischaemia.167-171 The Purkinje cells of the cerebellum, medium sized striatal neurons, and the CA1 pyramidal cells of the hippocampus are especially sensitive.172 Histological damage may be apparent after about 3 minutes of ischaemia for hippocampal neurons, but after about 30 minutes for striatal neurons.172 173 Furthermore, significant species and model differences in neuron vulnerabilities are possible.174 175 Despite these distinct cellular vulnerabilities, there is no evidence that during focal cerebral ischaemia they contribute to haemorrhage. Rather, this seems to be more dependent on the sensitivity of microvascular structures to ischaemia.
Haemorrhagic transformation in embolic stroke
Clinically, haemorrhagic transformation is manifest either by haemorrhagic infarction or parenchymal haematoma, or both.34 176 Okada et al 32 and others31 38 have shown that haemorrhagic infarction is more common than parenchymal haematoma in patients with carotid territory embolic stroke. Parenchymal haematomas seem to accompany acute carotid territory embolism more often in the setting of anticoagulant treatment.176-181 The longstanding concept that haemorrhagic transformation may result from arterial reperfusion is so far not supported by angiographic studies.11 12 35 182 Ogata et al have suggested that haemorrhage may also occur from collateral channels or other vascular sources.183 Based on these clinical relations, it may be postulated that arterial reperfusion is dangerous when the microvascular basal lamina and matrix are disrupted by processes initiated by acute closure of the feeding artery. Regions of haemorrhagic transformation are likely to be those in which the microvascular beds are most vulnerable. Processes which disrupt microvascular structure (for example, amyloid deposition) may increase this vulnerability.
Plasminogen activators, ischaemic injury, and haemorrhagic transformation
Plasminogen activation carries an inherent risk of haemorrhage by altering platelet plug framework, vascular permeability, and vascular basal lamina integrity at sites of injury.184-186Exogenous PAs might accelerate dissolution of the blood-brain barrier, microvascular basal lamina/ECM, and platelet-fibrin plugs thereby increasing oedema formation and the risk of haemorrhage. This hypothesis is consistent with the finding that the frequency of haemorrhagic transformation in the setting of rt-PA exposure increases with the time of intervention from symptom onset in patients with MCA occlusion,35 in whom intrinsically basal lamina/ECM dissolution increases with time from MCA occlusion.46
Five recent placebo controlled studies of intravenous PA infusion in acute ischaemic stroke highlight patient and study features which relate to the accentuated risk of haemorrhagic transformation (table1). Three separate projects examined the relative benefits of 1.5×106 IU streptokinase, the recommended dose for acute myocardial infarction.187 188 Hommel and colleagues of the Multicenter Acute Stroke Trial-Europe (MAST-E) reported a significantly higher incidence of symptomatic intracranial haemorrhage in the streptokinase group compared with placebo among patients treated within 6 hours of the onset of an MCA territory stroke.5 9 The significantly higher short term (10 day) mortality in the placebo group (18.1%), suggested that the increased risk of symptomatic parenchymal haemorrhage with streptokinase was due to the increased severity of strokes entered into that trial. The Australia Streptokinase (ASK) Trial, which randomised patients to intravenous placebo or streptokinase administered within 4 hours of acute ischaemic stroke, noted a significantly increased mortality among patients treated after 3 hours.4 6 The time from symptom onset to treatment did not seem to be relevant to the excess frequency of symptomatic haemorrhage in the streptokinase group. A third study, the Multicentre Acute Stroke Trial-Italy (MAST-I), which compared intravenous streptokinase ± aspirin and placebo within 6 hours of onset of symptoms, was terminated when interim analysis showed a significant excess of 10 day case fatalities associated with streptokinase ± aspirin, but most particularly when streptokinase was given with aspirin.7 8 The incidences of haemorrhagic transformation detectable by CT at 5 days and symptomatic cerebral haemorrhage during the hospital stay among patients treated with streptokinase ± aspirin significantly exceeded those in patients who received placebo.
ECASS, which tested the efficacy of rt-PA (alteplase) in acute ischaemic stroke applied within 6 hours of onset of symptom, did not show overall benefit in terms of disability outcome at 90 days.1 In that study, symptomatic brain haemorrhage was not defined as a primary outcome event. Intracranial haemorrhage was assessed on days 1 and 7 after treatment and categorised as haemorrhagic infarction or parenchymal haematoma.176Haemorrhagic infarction on day 1 did not affect mortality outcome, which was 12% in patients with haemorrhagic infarction and 15% in patients without haemorrhage. However, parenchymal haematoma on day 1 was associated with a 52% mortality (von Kummer et al, unpublished data). The severity of initial clinical deficit and the presence of ischaemic changes on CT were associated with risk of haemorrhagic infarction. Increasing age and treatment with rt-PA were related to the risk of parenchymal haematoma.37 In the rt-PA treated group, the risk of parenchymal haematoma increased with the extent of hypoattenuation on the initial CT.189In ECASS, the risk of cerebral haemorrhage was not apparently associated with the time from symptom onset to initiation of treatment, although the stroke population was heterogeneous with respect to aetiology.
The NINDS sponsored placebo controlled evaluation of rt-PA (alteplase) in patients with ischaemic stroke within 3 hours of onset of symptoms showed an 11% to 13% absolute improvement in best outcome using a composite of neurological outcome (NIHSS), two disability indices, and the Glasgow outcome score.2 Within an apparent equivocal effect on mortality was a significant increase in symptomatic intracerebral haemorrhage from 0.6% in the placebo group to 6.4% in the rt-PA group. One interpretation of those data is that the improvement in clinical outcome would have been substantially increased had the frequency of symptomatic haemorrhagic transformation been attenuated. This interpretation also applies to the ECASS experience.1
The excessive frequency of early symptomatic intracerebral haemorrhage in each trial could be attributed to the use of a plasminogen activator, and in at least two trials also to the severity of injury.1 5 In one trial, the impact of the injury on apparent risk may have been accentuated by an “excessive” dose of the streptokinase, and exposure of the injured microvascular bed to increased plasmin levels.5 In a subgroup of patients with evidence of ischaemic injury on the initial CT scan entered into ECASS, exposure to rt-PA significantly increased the frequency of symptomatic parenchymal haematoma within 24 hours (table 2). That experience implies that symptomatic haemorrhage complicates those regions with extensive early injury. Furthermore, a retrospective analysis of the NINDS dataset indicated that patients with severe strokes, or oedema, or mass effect on baseline CT had a higher frequency of intracerebral haemorrhage.3 A similar finding has been made in a placebo controlled study of intra-arterial infusion single chain urokinase plasminogen activator.25 These experiences are consistent with the postulate that the risk of symptomatic haemorrhage is a product of the depth and duration of ischaemic injury.
Outcomes in patients (n (%)) with no or small hypoattenuation v large hypoattenuation on initial CT
If it is true that the volume of brain tissue with irreversible ischaemic damage of microvessels is a dominant contributor to postischaemic cerebral haemorrhage, CT and MRI may identify patients with a potential increased risk of haemorrhage and mortality by exposure to PAs. Ueda et al have suggested that a threshold of rCBF reduction may be defined which is significantly associated with symptomatic intracerebral haemorrhage after PA exposure in acute stroke.190 From ECASS, evidence of tissue injury exceeding 33% of the MCA territory was associated with increased early symptomatic haemorrhage and mortality.189 The difficulty that CT and MRI findings during the first 2 hours may not be reliable because the disturbance of diffusion can be reversible191 or the CT may not display a lesion although oedema formation is ongoing temper this impression. However, if CT shows hypoattenuation during the first 2 hours after onset of symptoms, a territory with severe ischaemia and oedema is identified. In ECASS, 60 of 152 (39%) patients examined within the first two hours of symptom onset showed hypoattenuation on their CT (which exceeded 33% of the MCA territory in 14 patients (9%)).189 All patients with hypoattenuation on the initial CT independent of time of presentation showed necrosis on the follow up CT by 7 days (von Kummer, unpublished data).
Despite the apparent salutory effects of rt-PA in patients with ischaemic stroke treated within 3 hours of onset of symptoms,1 2 studies selecting and treating patients with ischaemic stroke with PAs based only on clinical criteria have shown an excess of symptomatic haemorrhage.1 2 4-8 This impression is supported by a recent meta-analysis.192 193Among smaller studies in which patients were selected on the basis of location of occlusion, with recanalisation as an outcome measure, the frequency of symptomatic haemorrhage did not differ significantly from those receiving placebo.194 195 In either case, if the hypothesis is correct that PAs may augment the dissolution of the microvascular basal lamina/ECM and thereby exacerbate haemorrhage based on their common characteristic to generate plasmin, then cerebral haemorrhage is likely to be a consequence. But the generation of plasmin necessary for thrombus lysis is intrinsic to PA activity and the consequences of plasmin generation on microvascular integrity are so far unalterable. So, attempts to decrease the clinical risk of plasminogen activator associated haemorrhagic transformation are necessary to improve overall outcome for patients and clinical studies.
The physician is currently left with applying strict criteria to select those patients with acute ischaemic stroke with attributes which may lower their intrinsic risk of haemorrhagic transformation. Specific characteristics which distinguish patients at risk for haemorrhage who receive a PA have not been resolved by experimental work and this is an important issue to be studied. Consideration of the roles that ischaemic microvascular injury may have in the processes of oedema formation and haemorrhagic transformation may guide that selection.
DEFINITION OF THE REGION OF ISCHAEMIC INJURY BY CT OR MRI
To reduce the inherent risk of intracerebral haemorrhage due to the PA, patients should be examined by CT or MRI to rule out primary haemorrhage as a cause of symptoms and to determine the volume of tissue which has had ischaemic injury. This is possible 2 hours after ictus, and in some patients even earlier. The critical volume of damaged tissue has yet to be determined. So far, a volume exceeding 33% of the MCA territory is associated with no benefit from PA and an increased risk for symptomatic haemorrhage.
DURATION OF INJURY
The designated time of onset of symptoms may underestimate the duration of ischaemic injury in some patients. Several uncontrolled and controlled studies have suggested that a decreased risk of haemorrhage is associated with shortened durations of ischaemic injury. Therefore, very early acquisition of patients to CT and potential treatment is required. Besides the need to initiate treatment very early, functional imaging methods to define the individual risk or extent of patients with sustained low rCBF on irreversibly damaged ischaemic tissue bear evaluation.
POTENTIAL CONTRIBUTORS TO MICROVASCULAR INJURY
The microvasculature is also a target for ischaemic and inflammatory injuries with the general consequences of increased permeability to plasma and circulating cells. Sustained exposure to raised blood pressure, diabetes mellitus, amyloid angiopathy, and comorbid inflammatory conditions (for example, vasculitis) are likely to augment the effects of ischaemia on the microvascular bed.
PLASMINOGEN ACTIVATORS AND THE MICROVASCULATURE
There is as yet no specific experimental data on the relative effects of various PAs on microvascular integrity. Whether certain PAs may promote additional injury is unknown. However, agents which initiate complement activation (for example, streptokinase)196 197 and affect endothelial cell integrity (for example, streptokinase and rt-PA),184 198could augment ischaemic microvascular injury. This argues that specific dose adjustment studies and prudent choice of agents should be an integral part of the testing of PAs in this setting. Furthermore, events which augment basal lamina and ECM dissolution (for example, increased inflammation) are likely to increase the risk of microvascular haemorrhage.
These issues are appropriate for further well conceived experimental work, but must also rigorously apply to the approved use of plasminogen activators in acute ischaemic stroke.
Conclusions
(1) Present experimental data allow the hypothesis that loss of microvascular integrity is one prerequisite for the development of haemorrhagic complications in focal cerebral ischaemia, with or without reperfusion.
(2) Loss of microvascular integrity may be increased by plasminogen activators although this has not been rigorously or prospectively examined.
(3) Because there is no currently available strategy to reduce or prevent loss of microvascular integrity during focal cerebral ischaemia, and therefore the intracerebral haemorrhagic complications, very careful patient selection based on clinical examination, CT, and perhaps MRI, must remain the current strategy.
Acknowledgments
We thank Marcia Filbert for her expert help in preparing this manuscript. This is publication number 11107-MEM from The Scripps Research Institute, La Jolla, CA, USA. This work was supported in part by grant RO1 NS 26945 of the National Institutes of Neurological Diseases and Stroke.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵