Article Text

Statistics from Altmetric.com
Machado-Joseph disease refers to autosomal dominant spinocerebellar degeneration, and the gene responsible for the disease exhibits an expanded trinucleotide CAG repeat in chromosome 14q32.1.1 Machado-Joseph disease has a wide range of clinical manifestations in addition to the cerebellar ataxia. The diverse disorders are characterised neuropathologically by the involvement of the pallidoluysian, dentatorubral, pontocerebellar, cochleocerebellar, and spinocerebellar systems, lower motor neurons, and dorsal root ganglia. Previous MRI studies disclosed only mild cerebellar and brain stem atrophy in Machado-Joseph disease.2 Our MRI examinations in 31 cases disclosed atrophy of the pons, middle, and superior cerebellar peduncles, and frontal and temporal lobes, together with fourth ventricular dilatation.3 A third of the cases displayed a hyperintense signal of the transverse pontine fibres, which had been found previously in patients with olivopontocerebellar atrophy.4Here, we report on a patient with Machado-Joseph disease with an abnormal pontine signal on MRI, nine months before death, and pathological findings of the necropsied brain.
A 46 year old man had been in good health until the age of 23 when he began to stagger and slur his speech. He showed progressive difficulty in walking and was bedridden at the age of 37. Eight years later, he was admitted to hospital because of dysphagia and dysarthria. His father had had Machado-Joseph disease and died at the age of 40.
The patient was 160 cm tall and weighed 34.8 kg. He was mentally inactive, but his orientation and memory seemed normal. Communication was difficult due to cerebellar ataxia, dystonia, and severe hypophonia. Abduction of the eyes was limited bilaterally, with impaired upward gaze, slow saccades, and gaze evoked horizontal nystagmus. He had bulging eyes and his speech was very hypophonic. Fine twitching movements were noted in his facial muscles. His limbs were hypotonic and wasted. Muscle strength was weak, and the legs were severely deformed due to contracture. Tendon reflexes in the upper limbs were exaggerated, but knee and ankle jerks were decreased. The Babinski reflex was positive. Dysmetria and dysdiadochokinesis were present in both arms. Dystonic posture was striking in his hands. CAG repeat lengths of the MJD1 gene were 77 and 19 (method described previously5). He died of acute interstitial pneumonia at the age of 46, nine months after MRI.
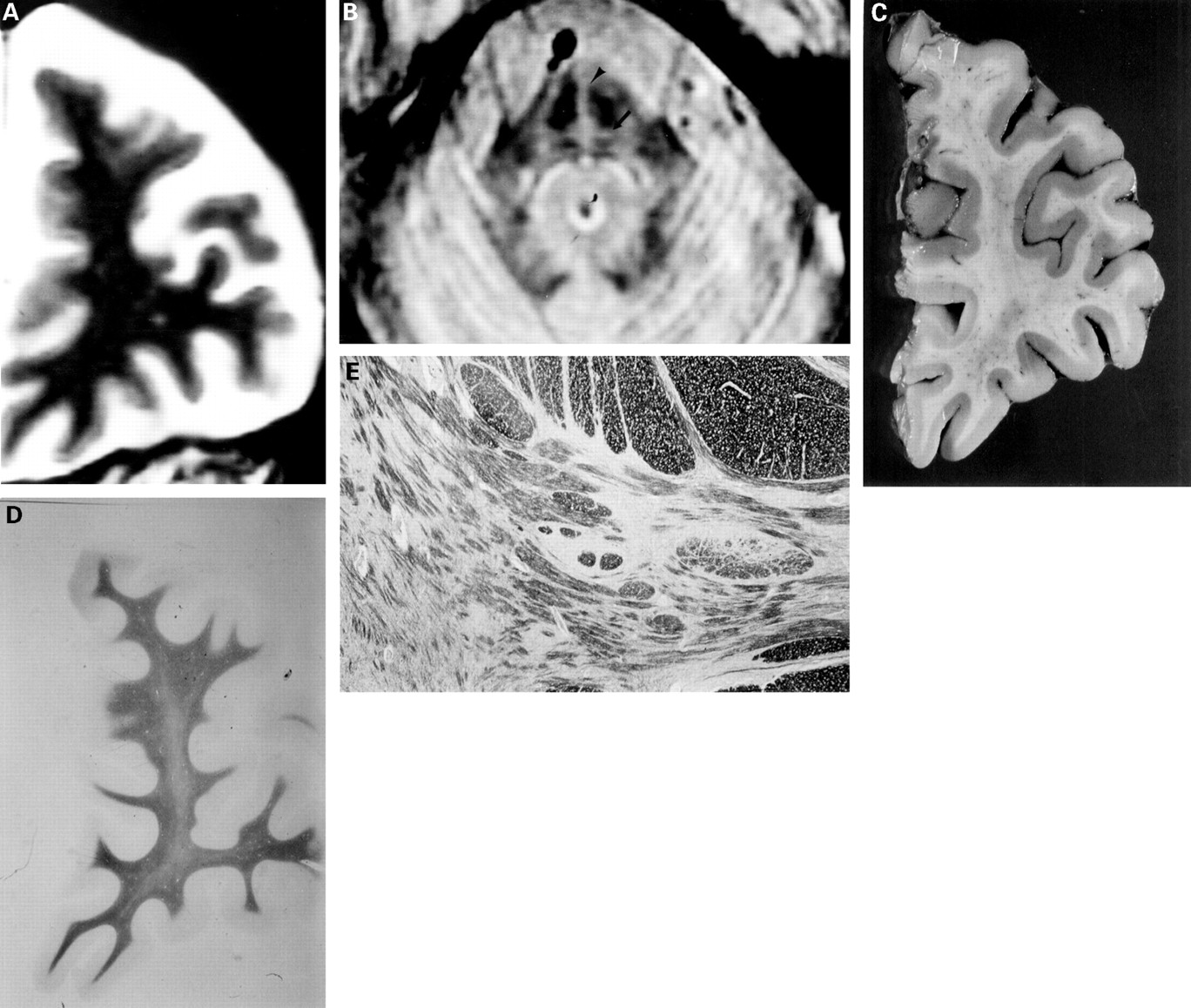
Brain MRI was carried out using Signa Advantage 1.5T. T1 weighted images (TR=450 ms, TE=19 ms), T2 weighted images (TR=3000 ms, TE=102 ms), and the first echo of long TR sequence images (TR=3000 s, TE=17 ms) were taken in the transaxial, coronal, and sagittal planes, at 5 mm thickness with a 2.5 mm gap. Brain MRI disclosed severe atrophy of the anterior lobe of the cerebellum, upper cerebellar peduncles or brain stem, and moderate atrophy of both middle cerebellar peduncles, bilateral frontal (figure A) or temporal cortex, as well as a hyperintense signal in transverse pontine fibres (figure B) and pontine midline on the first echo of the long TR sequence. The hyperintense signal in the pons was narrower and less intense than in most cases of olivopontocerebellar atrophy.

{kind=link}
(A) Coronal T2 weighted MRI (TR:3000 ms, TE:102 ms) showing moderate frontal atrophy. (B) Axial first echo of long TR sequences MRI (TR3000 ms, TE:17ms) showing hyperintense signal of transverse pontine fibres (arrow) and pontine midline (arrow head). (C) Section of the left frontal lobe fixed in formalin. The orbital gyrus shows moderate atrophy and the superior, middle, and inferior frontal gyri show mild atrophy. (D) Section of the left frontal lobe. The frontal white matter displays pallor of the myelin (Klüvar-Barrera stain, originally×2). (E) Medial part of the pons. The pontine nuclei and transverse fibres show pronounced atrophy with nerve cell loss, but without gliosis, demyelination, or change in neuronal density (KB stain, originally×10).
The weight of the fresh brain was 1320 g. Paraffin sections (7 μm) were made after fixing the specimens with formalin and staining with haematoxylin and eosin, Klüvar-Barrera, Bodian, and methenamine silver stains, and immunohistochemistry with antiglial fibrillary acidic protein and antiubiquitin antibodies. The brain atrophy was moderate in the frontal (figure C) and temporal lobes, and considerable in the pontine base and cerebellum. On microscopical examination, the cell architecture in the cerebral cortex was normal without senile plaques, neurofibrillary tangles, or inclusion bodies. The frontal white matter displayed pallor of the myelin (figure D). The globus pallidus showed mild neuronal cell loss and gliosis. The pontine nuclei, transverse fibres, and upper and middle cerebellar peduncles showed considerable atrophy associated with nerve cell loss, but without gliosis, demyelination, or change in neuronal density (figure E). The cerebellar white matter exhibited slight pallor on Kluvar-Barrera staining, and the dentate nuclei showed cell loss and gliosis. The cerebellar cortex was normal.
Characteristic findings on MRI in this case were the abnormal hyperintense signal of transverse pontine fibres and the mild frontal and temporal atrophy. The hyperintense signal in the pons of cases of olivopontocerebellar atrophy has been reported to reflect gliosis and myelin sheath loss along degenerated pontocerebellar fibres.4 Our previous MRI examinations have disclosed the abnormal signal of transverse pontine fibres in some cases of Machado-Joseph disease, whereas all cases of olivopontocerebellar atrophy show a more hyperintense and wider signal of transverse pontine fibres.3 The difference might arise from the lack of demyelination and gliosis in Machado-Joseph disease, and the decrease in the volume without change of the neuronal density in the pontine nuclei and transverse pontine fibres in this case.
The frontal atrophy found by MRI in 31 cases of Machado-Joseph disease was examined and found to be more accelerated than in age matched control subjects. The cell architecture of the frontal cortex was well preserved, but pallor of the myelin was noted in the frontal white matter. The frontal atrophy might be produced by demyelination in the deep white matter. The patient was mentally inactive, but his orientation and memory were not impaired, although a mental test was impossible because of his severe ataxia, dystonia, and hypophonia. Patients with Machado-Joseph disease do not usually show impaired cognitive function, despite mild to moderate frontal or temporal atrophy. A previous pathological study of Machado-Joseph disease disclosed similar frontal atrophy,6 in which the cerebral cortex was not greatly affected, but the frontal white matter volume was decreased.
In conclusion, the frontal atrophy commonly seen in Machado-Joseph disease may be caused by demyelination in the deep white matter and the hyperintense signal of transverse pontine fibres, visible on long TR sequences of MRI, is presumably due to the characteristic atrophy of the pons in Machado-Joseph disease.