Abstract
BACKGROUND AND PURPOSE: Changes of the major forebrain commissures in lissencephaly have not been systematically studied. We investigated the developmental differences of the commissures in patients with varying types of lissencephaly to determine whether specific commissural features may help in distinguishing lissencephaly phenotypes.
MATERIALS AND METHODS: MR imaging of 124 patients was retrospectively reviewed. Patients were classified as having cLIS, vLIS, and CBSC, according to cortical phenotype; few patients had genetic diagnoses. Abnormalities of the CC, AC, and HC were recorded, and the overall shape was regarded as hypogenetic, hypoplastic, dysmorphic, a thin flat callosal body with a vertical splenium, and a convex upward callosal body, compared with age-matched controls. Correlations between commissural characteristics and cortical patterns were analyzed by using the Monte Carlo simulation of χ2, extension to m × n table, and Fisher exact tests as appropriate (P < .05).
RESULTS: Patients were classified as having cLIS (57.4%), vLIS (38.4%), or CBSC (4.2%). The most common callosal developmental anomaly was hypogenesis with an absent rostrum, a small inferior genu, and a small splenium. An angled (90°) splenium was found to be significantly associated with cLIS, as was an excessively convex upward callosal body with VLDLR. ACC with an enlarged AC was found in all cases of ARX.
CONCLUSIONS: Specific patterns of the commissure anomalies were associated with certain types of lissencephaly. Callosal anomalies were more common than those of the AC or HC. Developmental variations of commissures may be useful as an imaging criterion in differentiating the groups of lissencephalies and may give insight into the processes causing these malformations.
Abbreviations
- AC
- anterior commissure
- ACC
- agenesis of the CC
- ARX
- lissencephaly with ARX mutation
- ARX-XLAG
- lissencephaly with ARX mutation, and an absent callosum, ambiguous genitalia
- CBSC
- cobblestone complex
- CC
- corpus callosum
- cLIS
- classic lissencephaly
- CMD
- congenital muscular dystrophies
- DCX
- lissencephaly with mutated DCX gene
- FCMD
- Fukuyama CMD
- FKRP
- lissencephaly with FKRP mutation
- HC
- hippocampal commissure
- LARGE
- lissencephaly with LARGE mutation
- LIS1
- lissencephaly with mutated LIS1 gene
- ND
- not determined
- (p)
- presumed
- POMT1, POMT2, and POMGnT1
- lissencephalies with POMT1, POMT2, POMGnT1 mutations
- RELN
- lissencephaly with RELN mutation
- TUBA1A
- lissencephaly with TUBA1A mutation
- VLDLR
- lissencephaly with VLDLR mutation
- vLIS
- variant lissencephaly
Cerebral corticogenesis includes 3 major steps: 1) cell proliferation, 2) cell migration and differentiation, and 3) cortical organization with formation of a mature cortex.1 Lissencephalies result from impaired migration of neurons from germinal matrices to the developing cerebral cortex.1–5 Disruption of normal neuronal migration can result from many different genetic and environmental disturbances; the result is usually a major brain malformation, including many types of heterotopia, lissencephaly, and cobblestone malformations (also called dystroglycanopathies because they result from abnormal glycosylation of α-dystroglycan).5–10
Lissencephalies were previously divided into 2 distinct groups: type I (classic) and type II (cobblestone). Recently, this classification has been expanded, and the groups have been separated on the basis of pathologic, imaging, and genetic features.5,9,11 A recently proposed classification includes the following: LIS1 and DCX causing cLIS; ARX, RELN, and VLDLR; a 2-layered cortex causing vLIS; and FCMD, FKRP, LARGE, POMT1, POMT2, and POMGnT1 causing CBSC.11 Almost all patients have neonatal hypotonia, epilepsy, and little developmental progress. The diagnosis is difficult when based only on clinical and laboratory findings, which are time-consuming and expensive. Fortunately, typical imaging features have been described in association with many mutations causing lissencephalies. This imaging phenotypic characterization may help in differentiating these malformations.6,12–34
Various anomalies of the major forebrain commissures, namely the CC, AC, and HC, are commonly observed in human lissencephalies. The aim of our study was to investigate and categorize the developmental differences of the forebrain commissures in a large group of patients encompassing all lissencephaly types (classic, variant, and cobblestone) to determine if specific features of size and shape may help to distinguish the lissencephaly phenotypes.
Materials and Methods
MR images or portions of MR images of 124 patients were retrospectively reviewed. The scans were obtained from the private teaching collection of the senior author, acquired during 23 years, from the teaching file of the radiology department at the University of California at San Francisco (searched by using “lissencephaly,” “agyria,” “pachygyria,” “band heterotopia,” “cobblestone malformation,” and “congenital muscular dystrophy” as key words) and from MR images reviewed for a grant studying the genetics of epilepsy. The MR images were included only if they were quality images through the cerebrum (to assess the type and severity of the cortical malformation) and through the cerebral commissures as determined by a consensus of the authors. All studies included T1-weighted and T2-weighted images with scans obtained in at least 2 planes (sagittal and axial). All cases were confirmed unanimously by the authors to show agyria, pachygyria, or cobblestone cortex in some part of the cerebrum. Because the cases were largely from radiologic referrals, clinical information was usually rather sparse (typically including only a short report describing developmental delay, seizures, and family history) or, in some cases, completely absent. Sixteen cases without midsagittal images, and 6 with only CT scans were excluded. Eight patients with Walker-Warburg syndrome who had severe hydrocephalus limiting assessment of some commissural structures were also excluded. The available data of the remaining 94 patients showed a mean age of 5.5 ± 7.5 years (range, 1 day to 32 years; median, 2 years) at the time of imaging. The group included 33 females, 44 males, and 17 with unavailable sex information.
MR images of 50 sex- and age-matched patients who were scanned for headaches, suspected macrocephaly, or epilepsy and interpreted as having normal findings by a board-certified neuroradiologist were reviewed as controls. The lack of abnormality was confirmed unanimously by all authors in all cases. These control MR images established the normal appearance of the forebrain commissures for comparison with our patients. All control MR images had sagittal T1-weighted images, axial T1-weighted images, and axial and coronal T2-weighted images with section thicknesses of 3–5 mm.
The authors reviewed the cases jointly with special attention paid to the cortical involvement. The patients were then classified to a presumed genotype on the basis of the imaging phenotype. Information about genotype was available only for a small percentage of our patients because most were studied before the genetic tests for lissencephalies (LIS1, DCX, TUBA1A, ARX, VLDLR, RELN) or cobblestone malformations (FCMD, FKRP, POMT1, POMT2, LARGE, POMGnT1) were available. These many patients were classified as presumed genotypes, (p)LIS1, (p)DCX, (p)ARX, (p)RELN, (p)VLDLR, if the imaging findings were highly suggestive of a specific gene mutation as detailed in a previous article.11 Cobblestone malformations were not subclassified because it has been well established that mutations of many glycosyltransferase enzyme genes can cause any of the clinical phenotypes; the clinicoradiologic phenotype is more dependent on the severity of the mutation (and presumably other still unknown causes) than on the gene that is mutated.10,33–35 All cases with ARX had a genetically proved diagnosis. All mutations (proved and presumed) were further grouped into the 3 main groups of cLIS, vLIS, and CBSC for comparison of imaging features.
We assessed the forebrain commissures, white matter volume, and myelination for each patient and compared these features with respect to the main groups. We compared the shapes of the CC in the midsagittal plane with those in the control group. The shape and size of each commissure was determined by visually comparing each subject with age-matched controls. Because there is significant variation in the size of the callosal components and of the commissures with age, as well as some variation among controls of the same age, only those commissures and callosal components judged unanimously by the 3 authors as significantly different from controls were graded as abnormal. The CC was categorized in the following manner.
Developmental Appearance
CC was classified according to the developmental appearance as: agenesis (absence of the entire CC), hypogenesis (absence of the rostrum associated with a small or absent splenium and a small or absent inferior genu) (Fig 1), hypoplasia (all parts formed but with decreased thickness, presumably due to reduced number of crossing axons, of any part of the CC), and dysmorphic (abnormal shape of any part of the CC, presumably due to abnormal development) (Fig 2).

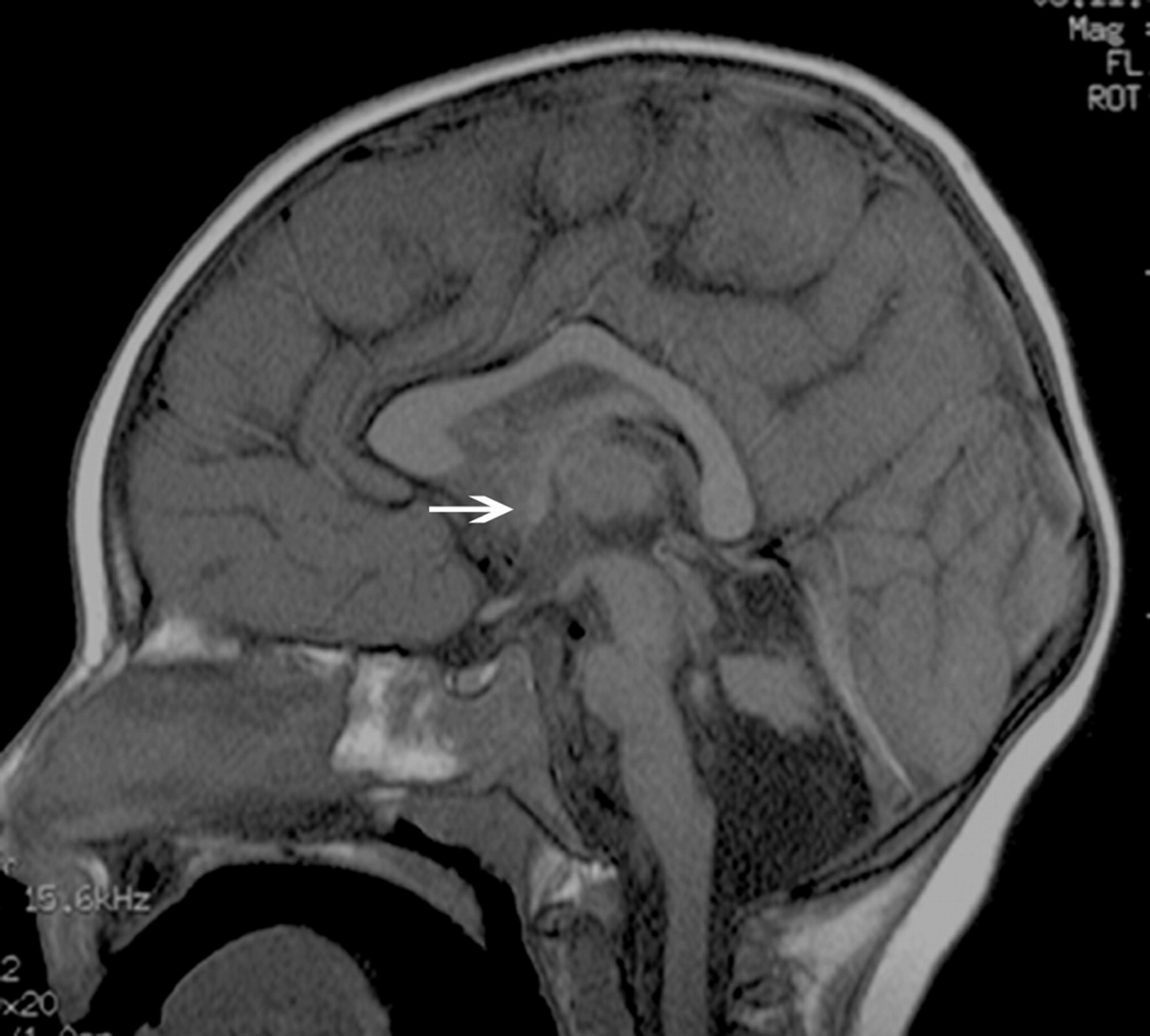
T1-weighted midsagittal image shows a hypogenetic CC in a patient with (p)LIS1. The rostrum is absent, the inferior part of the genu is small, and the splenium is thin. Note that splenium is also vertical; this was a common finding in classic lissencephalies. A normal AC is seen (arrow).

Midline sagittal T1-weighted image of a patient with a VLDLR; this is a dysmorphic CC with an abnormal shape and a convex upward callosal body configuration. The brain stem and vermis are hypoplastic, and the tentorium is displaced inferiorly. The AC is small (arrow).
Size of Parts (Rostrum, Genu, Body, and Splenium) and Overall Shape of the CC
Changes in size and shape of at least 1 part or another of the CC led us to observe mainly 2 recurrent overall callosal shapes in a large number of patients. Some had an absent rostrum, small inferior genu, and small splenium with a convex upward shape and a sharp angle at the level of the midcallosal body (Fig 3). Other patients had an absence of the rostrum, a small inferior genu, an abnormally thin flat callosal body, and a sharp angle (about 90°) between the body and a vertical splenium (Fig 4). These specific features were reminiscent of a hypogenetic dysmorphic CC.

Midsagittal T1-weighted image of a patient with the muscle-eye-brain disease phenotype of dystroglycanopathy. The splenium is thin, and the body is arched. This type of CC was classified as having a convex upward callosal body shape. Note the small pons, large tectum, and dysmorphic cerebellar vermis. The AC is normal (arrow); the patient has hydrocephalus.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The image of the (p)LIS case shows no rostrum, a small inferior genu, a flat corpus, and a vertically angled splenium. The case was classified as having a thin flat body and a vertical splenium shape. The CC has a sharp angle (approximately 90°) between the flat callosal body and the splenium. A small AC is seen (arrow).
The HC and AC were assessed on midsagittal T1- and coronal T1- or T2-weighted images. Coronal images were particularly helpful for the assessment of the HC because it connects the fornices and is usually largest immediately anterior and inferior to the callosal splenium. The size of the commissures was graded by visual assessment as small, normal, or enlarged compared with the control group.
The degree of myelination was reviewed and compared with the standards for the patient's age.35 For patients younger than 1 year of age, myelination >2 months below the standard for age was considered delayed. For infants between 1 and 2 years of age, myelination >3 months below standard was considered delayed. For children beyond their second birthday, only grossly reduced cerebral myelination was recorded as delayed. When a patient's age could not be determined or the images were insufficient for reviewing the myelination, it was recorded as nonassessed. White matter volume was visually assessed by the authors and considered normal or reduced. The proximity of cortical sulci to the lateral margins of the cerebral ventricles and the relative ratio of white matter to ventricular size after comparison with healthy controls were the criteria used for this assessment. Consensus was achieved among the authors for all cases; patients with questionable findings but close to healthy status were considered healthy.
Statistical comparisons of overall callosal shapes between classic and variant groups were carried out by the extension to m × n table, the Monte Carlo simulation of χ2, and Fisher exact tests as appropriate. ACC, hypogenesis, hypoplasia, a thin flat callosal body with a vertical splenium, a convex upward callosal body, and normal shape underwent statistical comparisons. A P value < .05 was considered significant. All statistical analyses were performed with the Statistical Package for the Social Sciences, Version 15.0 (SPSS, Chicago, Illinois).
Results
Among our 94 patients, 23 (24.46%) had a confirmed genetic diagnosis; 57.4% were classified as having cLIS (LIS1 = 3.7%, (p)LIS1 = 53.7%, DCX = 11.1%, (p)DCX = 29.6%, and 1 patient with no mutation), 38.4% as having vLIS (ND = 58.3%, RELN = 2.8%, (p)RELN = 5.5%, ARX = 11.1%, VLDLR = 5.5%, and (p)VLDLR = 16.6%), and 4.2% had CBCS (congenital muscular dystrophies−ND = 50% and muscle-eye-brain disease = 50%).
Most of the cases had developmental abnormalities of the forebrain commissures. The CC was more commonly affected than the AC and HC. The CC was most often hypogenetic and dysmorphic. This appearance of the CC clearly differed from that seen in the control group. Combining the proved and presumed same mutation groups, we applied statistics to evaluate group differences on the basis of the forebrain developmental abnormalities.
In cLIS, most of the patients with LIS1 (70.9%) had callosal hypogenesis (Fig 1), with a thin flat body and a vertical splenium shape in approximately half of the cases (48.4%) (Fig 4), which made a significant difference between LIS1 and other groups (P = .011). The AC and HC were normal in most of the cases. In patients with DCX, we found a hypogenetic CC (68.1%) with no specific dysmorphic shape. The AC and HC were mostly normal.
In the vLIS group, all patients with ARX had ACC with enlarged AC. The patients with the RELN mutation phenotype had a hypoplastic CC in all cases with an absent (33%) or small (66%) inferior genu. The AC was absent in 66%, while the HC was normal in all. The VLDLR phenotype was significantly associated with a hypogenetic convex upward CC body, which differed from the CC in the other groups (P = .011). The AC was predominantly small (62.5%), and the HC was mostly nonassessed (75%). The shape of the CC showed a significant difference between the cLIS and vLIS groups (P = .017). The patients with CBSC typically had an absent rostrum (75%), a thin or normal genu (75%), a thin or normal body (75%), and a thin splenium (50%). Because many of these patients had hydrocephalus or shunted hydrocephalus, the CC was often distorted and had a variable general appearance. Patients without severe hydrocephalus often had a smoothly arched callosal body (Fig 3). The AC and HC were normal in half of the cases.
Myelination was normal in 43.6% of all lissencephaly cases. Sixty-four percent of patients with cLIS and 18.9% of patients with vLIS showed normal myelination. Delayed myelination was found in half of the vLIS cases. The white matter volume was regarded as normal in only 6.4% of patients who had a normal CC. The remaining patients (93.6%) had differing severities of reduced white matter volume. All patients in the vLIS group showed reduced white matter volume.
Regarding the components of the CC, the rostrum was absent in 70.3% of cLIS and in 75% of vLIS cases. The genu was absent in only 7.4% of cLIS but in 30.6% of vLIS cases. A small inferior part of the genu was found in 59.3% of patients with cLIS and in 38.9% of those with vLIS. A thin flat body was seen more commonly in cLIS; it was frequently a convex upward callosal body shape in patients with vLIS (as stated above). When the splenium was not associated with previously described CC shapes, it appeared small in other cases. Statistical results are summarized in the Table.
Significant differences among the major types of lissencephalies for each structure and specific features
Discussion
This detailed review of a large number of MR images of patients with various types of lissencephaly showed developmental abnormalities of the major forebrain commissures in almost all patients; the CC was the commissure most commonly involved. This result is not surprising because the CC is composed of axons from cerebral cortical neurons that are abnormal in location and possibly in function in lissencephalies.36 Neurites from these abnormal neurons might be expected to experience errors in pathfinding through the many signals that normally guide them across the midline in the developing brain.37,38 Although many variations were seen, the most common anomaly consisted of an absent rostrum, a small inferior genu, and a small or abnormally shaped splenium: this configuration has been defined as hypogenetic.35,39 In addition, malformations secondary to specific mutations and presumed mutations seemed to have a characteristic overall shape of the CC.
The patients with LIS1 had predominantly callosal hypogenesis with a thin flat body and a vertical splenium shape (Fig 4), while the DCX phenotype was significantly associated with a hypogenetic CC without any specific overall CC shape. The ARX mutation with the XLAG phenotype is known to have callosal agenesis in nearly all cases (by definition). Therefore, finding ACC in our patients with ARX was expected. ACC was also found in other vLIS cases and could potentially be used to differentiate cLIS from vLIS. The patients with RELN mainly had a hypoplastic CC, while the CC in the patients with VLDLR was generally hypogenetic with a convex upward body shape (Fig 3). The patients with CBSC were included even though they have distinctive characteristic MR imaging features of the cerebral cortex and posterior fossa structures, because we aimed to explore the forebrain commissures within the full range of lissencephalies. We thus propose including our findings in the differential criteria of various lissencephaly MR imaging phenotypes.
The AC and the HC were assessed along with the CC because all are composed of axons from cortical neurons crossing through the cerebral midline.39 Thus, some developmental disturbances affecting the midline crossing callosal axons might be expected to involve the other commissures as well. Absence of the AC was common in patients with vLIS, and its enlargement was consistently detected in ARX (along with ACC). The significance of such enlargement is unclear. Regarding the HC, it was enlarged in some patients with vLIS, but this appearance was not helpful in differentiating the lissencephaly groups.
The developmental differences of forebrain commissures in various types of lissencephalic cortical malformations are likely related to the causes of these malformations, which means the effects of gene mutations on neurogenesis, neuronal migration, and neurite development. Axons from abnormal neurons or from neurons in abnormal locations are less likely to navigate normally across the midline via the cerebral commissures. Because the physiologic basis of neuronal and neurite development and migration is complex and incompletely understood to date, it is not helpful to speculate on the physiologic and molecular biologic differences that have resulted in the commissural anomalies described in this study. Nonetheless, a few general concepts seemed to emerge. Patients with the LIS1 pattern of cortical involvement (pachygyria gradient more severe posteriorly, involving the parietal and occipital lobes) had a more vertical splenium (Figs 1 and 4), while those with the DCX pattern (more severe anteriorly, involving the posterior frontal lobes) more often had normal or a slightly thin splenium. Patients with the vLIS pattern of lissencephalic cortical malformations, which, other than VLDLR, have less organized cortical histology than the cLIS patterns,8 had the most severe commissural anomalies with frequent ACC or CC hypoplasia. Although patients with VLDLR sometimes had nearly normal CCs, they more commonly had a relatively sharp upward angle of the midcallosal body, giving them the convex upward body shape. The cause of this angle is not clear, but if confirmed in other studies, it may be a useful imaging marker for this unusual disorder. Overall, our findings indicate that the appearance of the CC does not simply derive from the pattern of cortical involvement in lissencephalies but is likely the result of impaired neuronal and axonal migration at some step.
This study contains a number of assumptions and limitations. It is limited by the small number of cases with established genetic diagnoses. An attempt was made to increase this number by classifying studies as having a “presumed” genetic classification based on characteristic MR imaging features and their correlation with known pathology, such as cell-sparse zones, associated cerebellar anomalies, and cortical thickness. In a previous study, we showed that the genetically proved and presumed groups showed considerable homogeneity of MR imaging morphology.11 Thus, we believe that this classification as “presumed” mutations was both useful and justified. Many phenotypes of patients in the vLIS group were classified as ND because of a lack of total or part of the clinical information. Some of these were likely the 2-layered lissencephaly described by Forman (2005) or a result of cases of TUBA1A.9,40–43 We did not presume the diagnosis of TUBA1A in any patient because the phenotypes associated with this mutation were not disclosed at the time of the study and are still not fully established. From current literature, it appears that TUBA1A may result in either cLIS or vLIS patterns.40–43 It may be useful to reassess these cases in several years when these and other new causes of lissencephaly are better established. Despite these obvious deficiencies, this study has yielded important information for physicians and scientists interested in corticogenesis by adding to the knowledge of associated malformations.
Conclusions
Anomalies of the cerebral commissures are common in the lissencephalies and likely result from impairment of the migration of cortical neurons and of the navigation of their axons through the developing cerebrum. Looking at the commissures in lissencephalies may help to differentiate lissencephaly phenotypes and hopefully, in the future, genotypes. The information provided by our study will hopefully lead to further studies of the molecular disorders involved in brain anomalies associated with agyria and pachygyria and, ultimately, a better understanding of the mechanisms of normal and abnormal brain development.
Acknowledgments
We acknowledge Christopher Walsh, MD, and William Dobyns, MD, who referred many of the MR imaging scans reviewed for this study.
Footnotes
-
This work was supported in part by NIH grant R01 NS035129.
Indicates open access to non-subscribers at www.ajnr.org
References
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- Received August 21, 2009.
- Accepted after revision March 22, 2010.
- Copyright © American Society of Neuroradiology