
Abstract
BACKGROUND AND PURPOSE: Reports of MR imaging in hypothalamic hamartomas associated with epilepsy are few, and the number of patients studied is small. We aimed to detail the relationship of hypothalamic hamartomas to surrounding structures, to determine the frequency and nature of associated abnormalities, and to gain insight into mechanisms of epileptogenesis.
METHODS: We systematically examined MR imaging studies of 72 patients with hypothalamic hamartoma and refractory epilepsy (patient age, 22 months to 31 years). A dedicated imaging protocol was used in 38 cases. Proton MR spectroscopy of the hypothalamic hamartoma was performed for 19 patients and compared with the metabolite profile of the thalamus in 10 normal children and the frontal lobe in 10 normal adults.
RESULTS: Compared with normal gray matter, hypothalamic hamartomas were hyperintense on T2-weighted images (93%), hypointense on T1-weighted images (74%), and had reduced N-acetylaspartate and increased myoinositol content shown by MR spectroscopy. Hypothalamic hamartomas always involved the mammillary region of the hypothalamus, with attachment to one or both mammillary bodies. Intrahypothalamic extension (noted in 97%) tended to displace the postcommissural fornix and hypothalamic gray matter anterolaterally, such that the hypothalamic hamartomas nestled between the fornix, the mammillary body, and the mammillothalamic tract. Larger hamartoma size was associated with central precocious puberty. Associated findings of questionable epileptic significance included anterior temporal white matter signal intensity abnormalities (16%) and arachnoid cysts (6%). Malformations of cortical development were observed in only two patients, and hippocampal sclerosis was not observed.
CONCLUSIONS: Hypothalamic hamartomas can be readily distinguished from normal hypothalamic gray and adjacent myelinated fiber tracts, best appreciated on thin T2-weighted images. MR imaging and spectroscopy suggest reduced neuronal density and relative gliosis compared with normal gray matter. Associated epileptogenic lesions are rare, supporting the view that the hypothalamic hamartoma alone is responsible for the typical clinical features of the syndrome. The intimate relationship to the mammillary body, fornix, and mammillothalamic tract suggests a role for these structures in epileptogenesis associated with hypothalamic hamartomas.
Hypothalamic hamartomas are developmental malformations associated with central precocious puberty and gelastic (laughing) seizures (1). Evidence for the role of hypothalamic hamartomas in the production of gelastic seizures has been found only during the last decade, with stereotactic depth recording of seizure onset within hypothalamic hamartomas (2–5), production of typical gelastic seizures by depth electrode stimulation of hypothalamic hamartomas (4, 6) and ictal single photon emission CT studies showing hypothalamic hamartoma hyperperfusion (6–8). Surgical treatment of hypothalamic hamartomas for intractable epilepsy is increasingly advocated, and a number of approaches have been described, including microsurgical techniques (8–11), stereotactic RF coagulation (6, 12), and radiosurgery (13, 14). Consideration of the anatomy of the lesion in relation to the surrounding structures is essential when planning surgery (8–10, 14) and in anticipating potential neurologic and endocrine complications of treatment (15, 16); optimal radiologic display and detailed study of the intrahypothalamic relationships are therefore desirable.
The heavily myelinated white matter tracts of the hypothalamus (postcommissural fornices and mammillothalamic tracts) are readily visualized with MR imaging of cadaver brains (17), healthy volunteers, and patients with tumors involving the hypothalamus (18). These tracts may play a role in seizure propagation (19) and in the evolution of gelastic seizures to symptomatic generalized epilepsy in patients with hypothalamic hamartoma (20), but the MR anatomy of these tracts in relation to hypothalamic hamartomas has not been described and descriptions of mammillary body involvement are limited to the presence or absence of an attachment (21).
Previous reports of MR imaging findings of patients with hypothalamic hamartoma and epilepsy are either single case studies or small series, some supplemented by a review of the literature (21, 22). The larger published surgical series deal mainly with the results of hypothalamic hamartoma resection in cases of refractory epilepsy (9, 10). In the literature, controversy exists regarding the frequency and epileptic significance of malformations of cortical development associated with hypothalamic hamartomas (1, 23). We herein report the MR imaging appearance and spectroscopic findings of epileptogenic hypothalamic hamartomas, the relationship of hypothalamic hamartomas to hypothalamic white matter tracts and myelinated nuclei, and the nature and frequency of associated structural brain abnormalities.
Methods
We examined the MR imaging studies of 72 patients with hypothalamic hamartoma and epilepsy. Forty-three patients underwent formal assessment at this center, 37 of whom subsequently underwent surgery; histopathologic examination of the resected lesions in those cases revealed glial cells and neurons of normal appearance, in keeping with hamartoma. The remaining patients had MR images and clinical details provided by the referrer. The study was approved by the Ethics in Human Research Committees of the Royal Children’s Hospital and by the Human Ethics Committee of the Austin and Repatriation Medical Centre.
Imaging Protocol
Thirty-eight patients underwent MR imaging at this center. A 1.5-T clinical MR imaging system (Echospeed; GE Medical Systems, Milwaukee, WI) was used with software versions 8.35M and 9.0 and a standard quadrature transmit-receive head coil. The protocol consisted of sagittal view fast spin-echo (section thickness, 3.5 mm; intersection gap, 0.1 mm), axial view fast spin-echo (section thickness, 3 mm; intersection gap, 0.5 mm), sagittal view conventional spin-echo T1 (section thickness, 2.5 mm; intersection gap, 0.1 mm), and sagittal view 3D inversion-prepared fast spoiled gradient-echo (contiguous 0.6-mm sections) imaging. Dedicated coronal view images targeted to the hippocampus and anterior temporal lobes were acquired perpendicular to the long axis of the hippocampus and included fast spin-echo (contiguous 3-mm sections) and fluid-attenuated inversion recovery (contiguous 4-mm sections) images. 2D reconstructions of 3D fast spoiled gradient-echo volumes in sagittal and coronal planes (contiguous 2.5-mm sections) were obtained for review purposes.
For 34 patients, only studies obtained at other centers were reviewed. These varied in the number, type, and quality of sequences and planes of imaging performed; therefore, inclusion criteria were determined as follows: for hypothalamic hamartoma review, we required T1-weighted or gradient-echo or inversion recovery sequences, T2-weighted or proton density–weighted or fluid-attenuated inversion recovery sequences, and images obtained in two orthogonal planes, with section thickness of not more than 5 mm and intersection gap of not more than 1.5 mm in each sequence; for review of associated abnormalities, we additionally required whole brain images of two different sequences (as above) and coronal plus one other plane of imaging. Three additional patients were excluded from the study because of poor quality MR images. Contrast-enhanced studies were reviewed for 55 of 72 patients, but contrast medium was not administered as part of the imaging protocol at our center.
MR Imaging Review
A neuroradiologist (L.T.C.) and a neurologist (J.L.F.) together systematically examined all MR images by using a data entry form. Information obtained for each patient included number of studies, details of sequences performed, patient age, and time intervals between multiple studies when performed. Hamartoma data sought by visual inspection included signal intensity characteristics across sequences, position of lesion in relation to floor of third ventricle (intraventricular and interpeduncular extension), hypothalamic regions occupied by hamartoma in midsagittal section (mammillary, tuberal, supraoptic, preoptic), relation to optic chiasm and pituitary stalk, presence of asymmetric attachment to hypothalamus proper with description of this by category (right-sided, predominantly right-sided, bilateral, predominantly left-sided, left-sided), contact by either fornix with hamartoma and any fornix displacement, and contact by either mammillary body with hamartoma and any mammillary body displacement or deformity. Maximum diameter of the hypothalamic hamartoma in any plane was measured to the nearest millimeter with viewing software (eFilm; eFilm Medical, Toronto, Canada), for those patients whose studies were available in digital format, or by reference to calibration scales on hard copy films after digitization of images. Hypothalamic hamartoma size, location, and involvement of neuroendocrine structures were compared for patients with and without precocious puberty by using the two-sample t test for hypothalamic hamartoma size and Fisher’s exact test for categorical data; significance level was set at P < .05. Whole brain visual inspection included lobar organization and gyral arrangement; hippocampal size, shape, and signal intensity; anterior temporal white matter signal intensity and gray-white interface; appearance of white matter generally and of specific fiber tracts (anterior commissure, corpus callosum); ventricular, basal ganglia, brain stem, cerebellar, and major vessel appearance; and presence of associated developmental anomalies, including malformations of cortical development.
MR Spectroscopy
Short echo (30 ms) MR spectroscopy was performed at the same time as structural MR imaging for 19 patients (age range, 2–21 years; average age, 11.8 ± 5.7 years) who had not previously undergone hypothalamic hamartoma surgical procedures. A point-resolved spectroscopy sequence with two chemical shift selective imaging pulses for water suppression was used. Spectra were acquired with 128 transients of two k data points over a frequency width of 5000 Hz by using TR 1500. A single voxel was placed in the hypothalamic hamartoma, the size of which was tailored to fit the lesion. Two regions in normal volunteers were used for comparison. Using the same clinical MR imaging system and spectroscopy protocol (1500/30 [TR/TE]; transients, 128), a 1 × 1 × 1.2 cm voxel was placed in the left thalamus of 10 children (age range, 5–17 years; average age, 11.3 ± 4.6 years) who had normal structural MR imaging findings, normal cognitive development, and no history of seizures. On a different 1.5-T clinical MR imaging system (Echospeed GE-LX2, GE Medical Systems) with a similar point-resolved spectroscopy sequence (1500/35; transients, 128), a 2 × 2 × 2 cm voxel (including both gray and white matter) was placed in the right frontal lobe of 10 healthy adult volunteers with normal structural MR imaging findings.
Metabolite concentrations were determined for each region of interest by using computer software (LCModel) (24) and a library of reference spectra in a basis set recorded by the imaging unit manufacturer and calibrated with a 50 mmol/L N-acetylaspartate solution. Spectra were analyzed to provide information regarding total N-acetylaspartate content, choline-containing compounds, creatine plus phosphocreatine, glutamine plus glutamate, and myoinositol. Results are presented in institutional units (approximating millimolar concentration) based on this process. Comparison of metabolite concentrations between hypothalamic hamartomas and normal tissues was conducted by using one-way analysis of variance and a post hoc Bonferroni correction for multiple comparisons. The significance level was set at P < .05.
Results
Clinical Features
All patients had seizures that were refractory to antiepileptic medications. Seizure onset (known for 64 patients) was from birth to 16 years (median, 3 months). Patient age was 22 months to 31 years at the time of the last MR imaging study (mean, 12 years). A history of precocious puberty was present in 28 patients, absent in 39, and unknown for five. Two patients with multi-system malformations of the Pallister-Hall syndrome phenotype were excluded from the study because of poor quality MR images. Two included patients had postaxial polydactyly but did not meet established clinical criteria for Pallister-Hall syndrome (25). No other patients had malformations outside the CNS.
Twenty-eight patients had previously undergone neurosurgical procedures; 19 underwent procedures directed toward the hamartoma, and 13 underwent other procedures (four underwent both hamartoma and other neurosurgical procedures). Regarding hamartoma analysis, preoperative studies were available for seven of 19 patients who had undergone previous hypothalamic hamartoma procedures; for the other 12 patients, procedures undergone included partial resection (five patients), biopsy only (four), RF coagulation (two), and stereotactic radiosurgery (one). Regarding review of associated brain abnormalities, preoperative studies were available for only two of 13 patients who had undergone previous non-hamartoma neurosurgical procedures; for the other 11 patients, procedures undergone included temporal lobectomy (four patients), corpus callosotomy (two), drainage of arachnoid cyst (two), frontoparietal corticectomy with additional multiple subpial transactions (one), bilateral ventriculoperitoneal shunt insertion (one), and brain biopsy (one). In all, 19 patients had only postoperative studies.
Hamartoma Properties
Images of 72 patients were included in this analysis. More than one study was reviewed for 49 patients, and the interval between first and last studies reviewed was 4 months to 13 years (average, 41 months). No hypothalamic hamartomas increased in size or changed appreciably in appearance during the interval between studies. The youngest patient studied was 3 months old and had seizure onset from day one; during 4 years of follow-up, the hypothalamic hamartoma decreased in size from 30 × 21 mm to 21 × 14 mm. For the 55 cases in which contrast medium was administered, no enhancement of the lesions was observed. Signal intensity of the hypothalamic hamartoma was increased (relative to cortical and deep gray matter) on T2-weighted images of 67 patients (93%) and was slightly decreased on T1-weighted images of 53 patients (74%); for patients who underwent both conventional spin-echo and fast spoiled gradient-echo imaging, hamartoma hypointensity, when present, was better appreciated on the fast spoiled gradient-echo images and rarely apparent on conventional spin-echo images. Some heterogeneity of solid tissue signal intensity was observed in seven cases (10%). Proton density–weighted or fluid-attenuated inversion recovery imaging was performed in 67 cases, 58 (87%) of which showed relatively high signal intensity compared with gray matter.
The size of hypothalamic hamartomas ranged from 8 to 42 mm in maximum diameter (mean, 19 mm; SD, 8 mm). The position of the lesions in relation to the usual location of the floor of the third ventricle and the hypothalamic regions varied with the size of the lesion (Fig 1). The smallest were entirely or predominantly intraventricular, whereas larger lesions were both intraventricular and interpeduncular. Splaying of one or both cerebral peduncles was present in the largest hamartomas, and eccentric positioning of the basilar artery was observed in two patients. Only two hypothalamic hamartomas were entirely below the floor of the third ventricle, the smaller of which (Fig 2) represents a rare exception to the observation made by Arita et al (22) that the parahypothalamic type of hypothalamic hamartoma is not associated with seizures. The midsagittal view images showed that all 72 hamartomas involved the mammillary region of the hypothalamus and all except six encroached onto the tuber cinereum; 16 large lesions (22%) extended rostrally to abut the optic chiasm, 12 of which also extended to the lamina terminalis in the preoptic region. Two large hypothalamic hamartomas had one or more cystic components with signal intensity similar to that of CSF within the lesion (Fig 3).

Reformatted T1-weighted (fast spoiled gradient-echo) midsagittal and tilted coronal view MR images of three patients show the range of hypothalamic hamartoma sizes and their locations with respect to regions of the hypothalamus. The smaller lesion (A) is wholly intraventricular and confined to the mammillary region, without encroaching onto the tuber cinereum. The larger lesion (C) extends to the lamina terminalis in the preoptic region, abuts the optic chiasm, and displaces the pituitary stalk. Two hamartomas (A,C) are slightly hypointense relative to cortical and deep gray matter.

T1-weighted coronal view and T2-weighted midsagittal view MR images show a rare example of an epileptogenic hamartoma located entirely below the floor of the third ventricle. The attachment of this small (6 × 5 × 8 mm diameter) hamartoma is narrowed at the level of the tuber cinereum, and a connection to the right mammillary body can be seen in the sagittal view image, but no distortion of third ventricular outline. This 12-year-old female patient had episodes of crying and laughing from 3 months of age, with increased seizure frequency and associated blank staring, disorientation, and fearfulness from age 9 years; interictal EEG findings were initially normal, but the patient later developed right occipital epileptiform discharges during sleep.
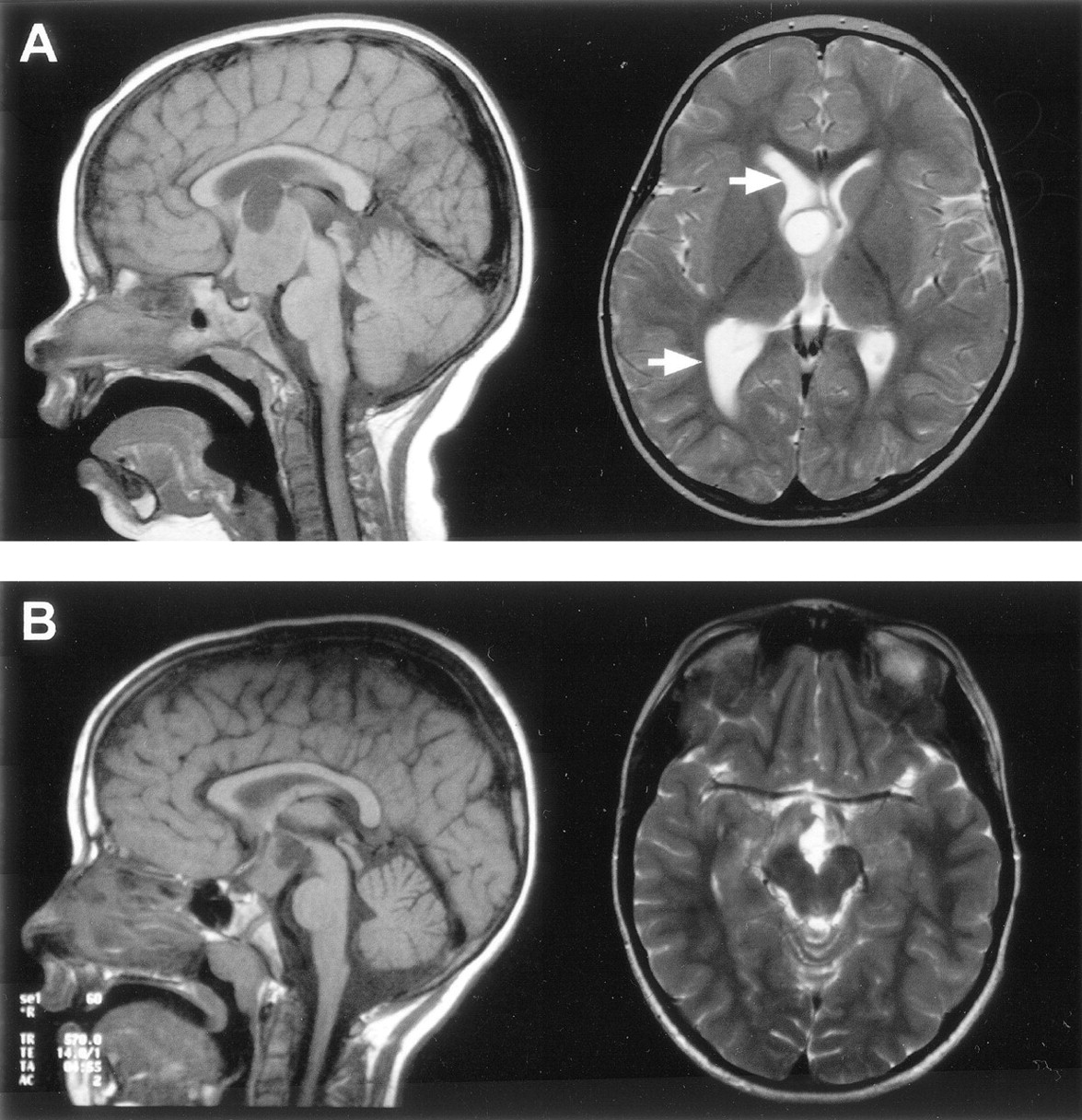
T1-weighted midsagittal view and T2-weighted axial view MR images show the cystic components of two large hamartomas with interpeduncular, prepontine extension.
A, In the first case, the cyst extends above the roof of the third ventricle and compresses the foramen of Monro on the right, resulting in dilation of the lateral ventricle (arrows).
B, In the second case, the cyst is within and extends below the third ventricle.
Fifteen hamartomas were in contact with the pituitary stalk, six of which displaced the stalk anteriorly, such that the stalk draped over the anterior border of the lesion. The pituitary stalk could not be identified in an additional four patients whose hypothalamic hamartomas had preoptic extension. Comparisons of hamartoma size, location, and involvement of neuroendocrine structures in patients with and without precocious puberty (known for 67 patients) are shown in Table 1. Features that were significantly associated with precocious puberty included larger hamartoma size (P=.004) and contact with the pituitary stalk (P=.01). Absence of tuberal involvement was predictive of normal pubertal development (P=.04).
Comparison of hypothalamic hamartoma size and other features in patients with and without precocious puberty
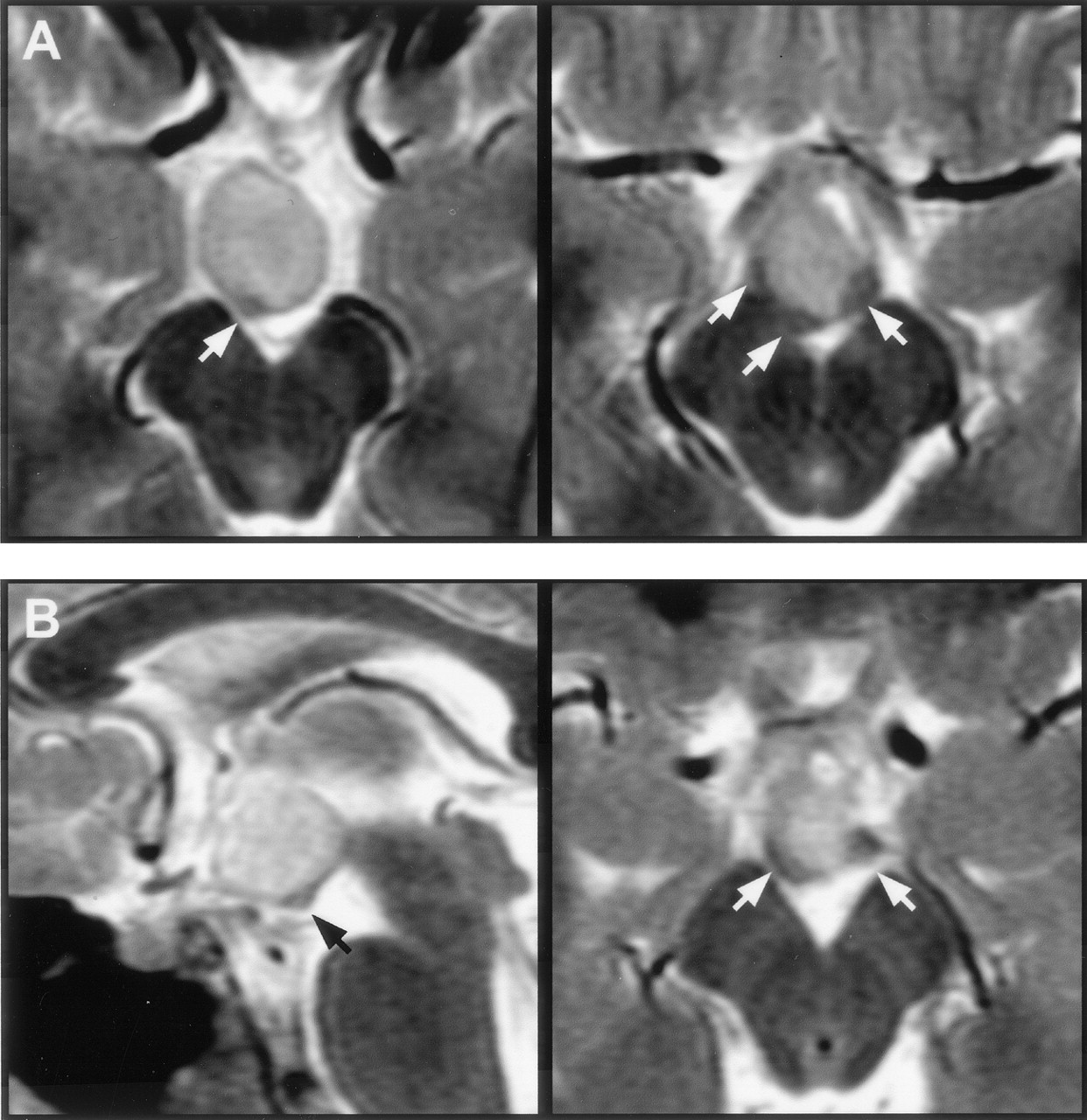
Attachment of the hamartoma to the hypothalamus proper was best appreciated on coronal view images. It was bilateral in 27 patients (37%) and predominantly or wholly unilateral in 45 (63%) (Fig 4). Of those identified as having bilateral attachment, 11 (41%) were noted to have greater involvement of one side. Axial and coronal view T2-weighted fast spin-echo images provided the greatest contrast and resolution for identifying intrahypothalamic relationships of the hamartoma. Both postcommissural fornices were identified within the hypothalamus of 66 patients (92%). Where intraventricular extension of the lesion was present (all except two cases), the hypothalamic hamartoma typically displaced the fornix and hypothalamic gray matter anterolaterally on the side of greater hypothalamic attachment. Fornix contact with the hamartoma was unilateral in 44 patients and bilateral in 24; fornix displacement was unilateral in 33 patients and bilateral in 12. According to an accepted definition of the boundary between the tuberal and mammillary regions of the hypothalamus (26), the intrahypothalamic part of the hamartomas therefore occupied mainly the mammillary region, nestled between the postcommissural fornix anteriorly, the mammillary body inferiorly, and the mammillothalamic tract posteriorly (Fig 5). The mammillothalamic tracts were not displaced by the hypothalamic hamartomas despite their intimate relation.

T2-weighted tilted coronal view image shows an example of a hamartoma with unilateral attachment to the hypothalamus (arrow).

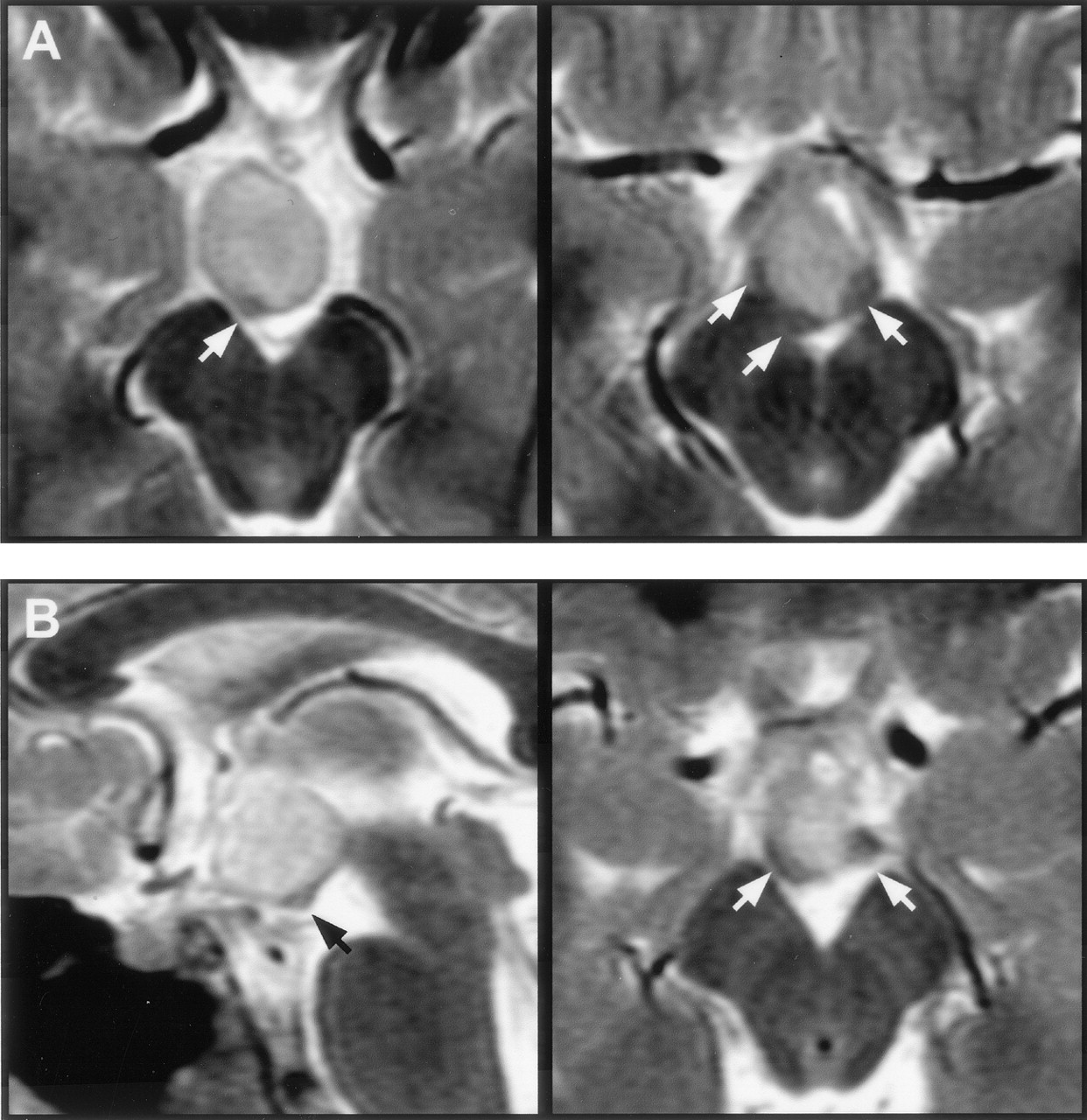
T2-weighted MR images with magnified views (insets) show the relation of the intra-hypothalamic component of the hamartoma to myelinated fiber tracts and gray matter of the hypothalamus in two patients.
A, Axial section shows anterolateral displacement of the postcommissural fornix on the left when compared with the unaffected right fornix, plus increased T2 signal intensity of the hamartoma relative to the hypothalamus.
B, Parasagittal section obtained at the level of the fornix shows the hamartoma between the postcommissural fornix anteriorly and the mammillothalamic tract posteriorly. Labeled are the postcommissural fornices (black arrows), mammillothalamic tracts (white arrows), and anterior commissure (arrowhead).
Mammillary bodies, or myelinated tissue on the floor of the third ventricle having the appearance of malformed mammillary bodies, could be identified in all except three patients. Displacement or distortion of mammillary tissue was unilateral in 47 patients (68%) and bilateral in 13 (19%). Distorted mammillary tissue could be identified as a thin, medially concave crescent applied to the posterolateral surface of the hamartoma (Fig 6). In 10 patients, mammillary tissue could be identified only unilaterally, and in eight of these patients, there was predominantly or wholly unilateral attachment of the hypothalamic hamartoma to the side on which no identifiable mammillary tissue was present.
Magnified T2-weighted images of two patients at the level of the mammillary bodies.
A, Axial sections in the first case show inferior displacement and crescent-shaped distortion of the right mammillary body with less marked abnormality on the right.
B, Midsagittal section in the second case shows inferior displacement of the right mammillary body. Axial section is similar in appearance to that shown in A. Labeled are the mammillary bodies (arrows).
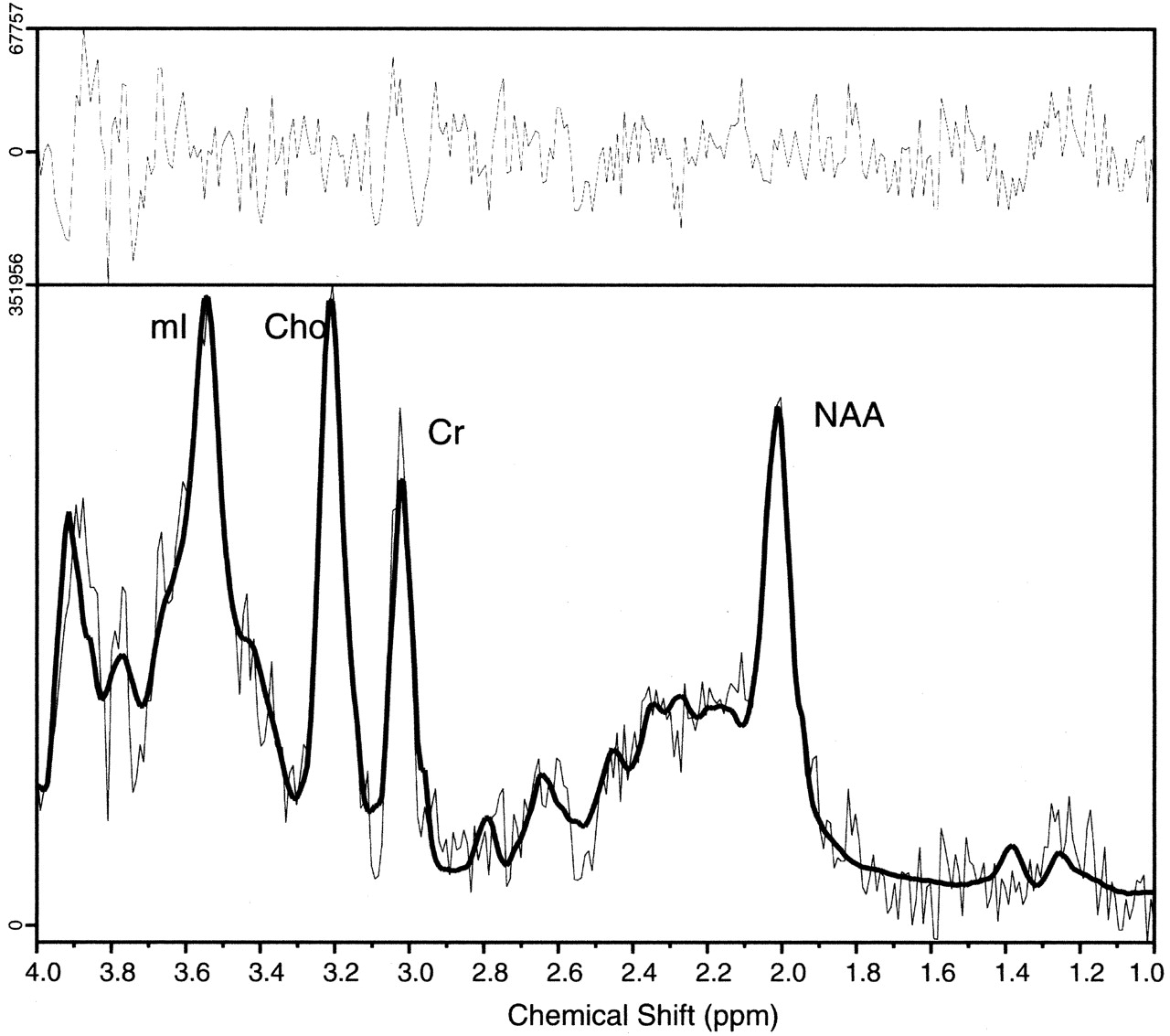
Comparison of proton MR spectra between hypothalamic hamartomas (example in Fig7) and normal tissues revealed significant differences in myoinositol (P = .0003) and N-acetylaspartate (P < .0001) concentrations but no differences in choline, creatine, or glutamine plus glutamate concentrations. Post hoc analysis (Table 2) revealed that myoinositol was elevated in hypothalamic hamartomas when compared with either thalamus or frontal lobe, whereas N-acetylaspartate was reduced by comparison.

Single voxel MR spectrum recorded from a hypothalamic hamartoma showing (from left to right) signal intensity from myoinositol (mI, elevated), total choline (Cho), creatine/phosphocreatine (Cr), and N-acetylaspartate (NAA, reduced) content. The spectrum was recorded from a 4-cm3 region tailored to the hypothalamic hamartomas by using a point-resolved spectroscopy sequence with 3000/30 (TR/TE). The solid line overlaid on the spectrum shows the fitted spectrum produced by LCModel (24), and the upper panel shows the residual of the fit.
Metabolite concentrations (mean ± SD) in hypothalamic hamartomas of patients compared with the thalamus and frontal lobe of control volunteers
Associated Findings
The whole brain imaging of 69 patients was included in this analysis. Acquired surgical abnormalities were noted in 28 patients (described above) and are not considered further. Seventeen patients (25%) had other abnormalities (Table 3; Figs 8–10). The most frequent finding, noted in 11 patients (16%), was increased T2-weighted signal intensity of the anterior temporal lobe white matter with loss of anterior temporal gray-white matter differentiation (Fig 8); for those with unilateral abnormality (seven patients), this was ipsilateral to the side of predominant hypothalamic hamartoma attachment in every case. Hippocampal sclerosis was not seen and was also not reported as a surgical or histologic finding for the four patients who underwent temporal lobectomy.
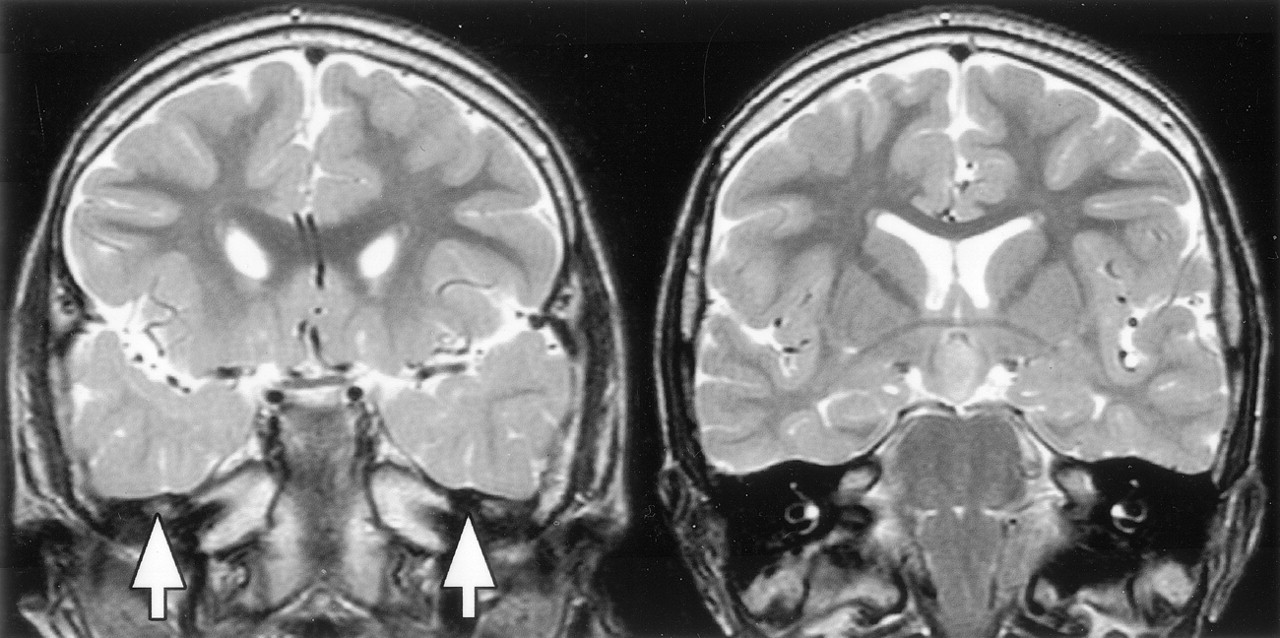
T2-weighted tilted coronal view MR images show an example of bilateral white matter signal intensity abnormality and decreased gray-white matter differentiation in the anterior temporal lobes (image on the left), with normal signal intensity and differentiation in the posterior temporal lobes at the level of the hypothalamic hamartomas (image on the right).
Associated findings in 69 patients with hypothalamic hamartoma and epilepsy
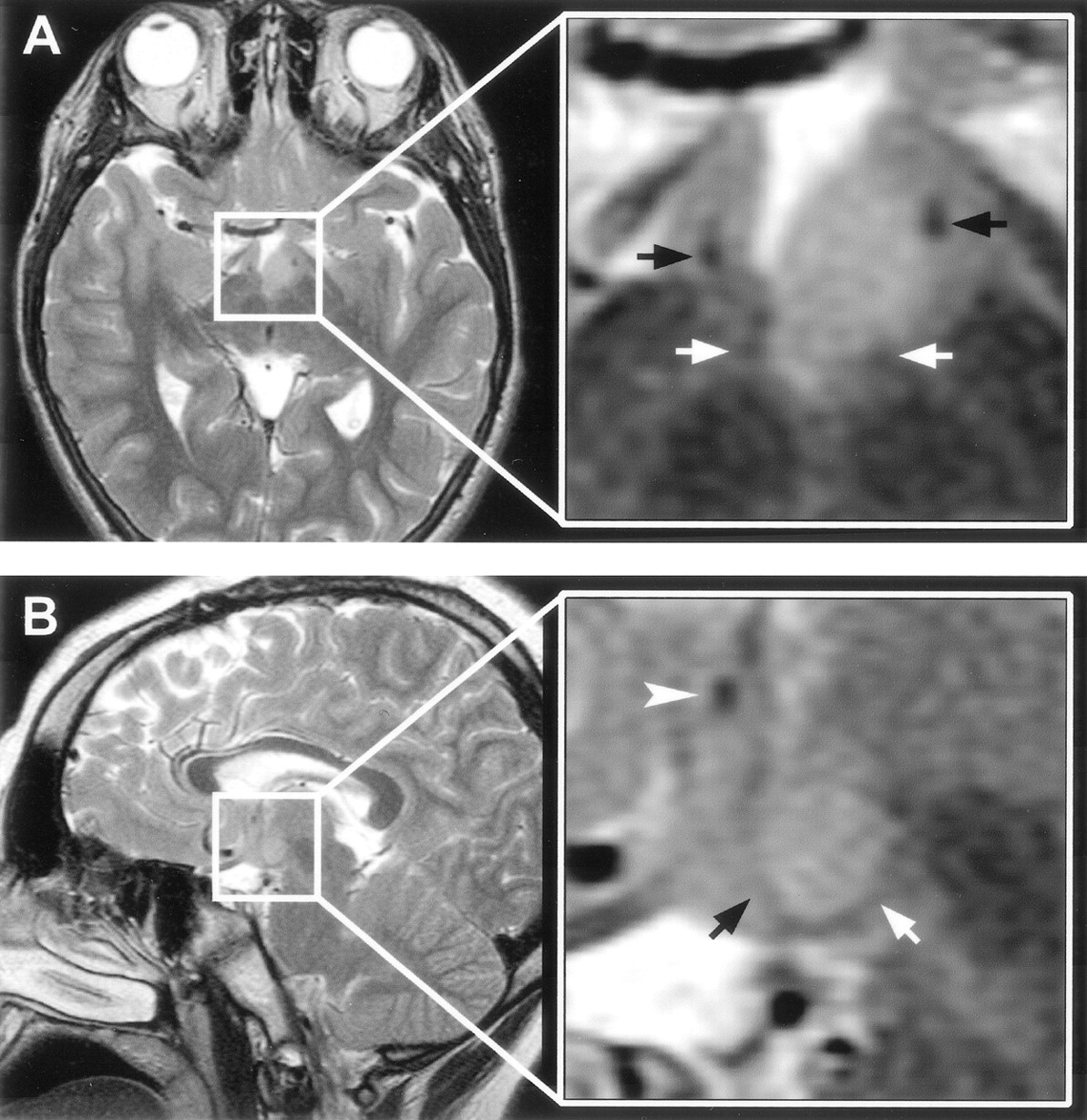
Malformations of cortical development were observed in only two cases (Fig 10). In the first case, periventricular nodular heterotopia and focal dilation of the left lateral ventricular trigone was associated with mesial occipital and temporal-parietal-occipital convexity cortical thickening and with abnormal gyral configuration, consistent with a transmantle, proliferative-type dysplasia (27). In the second case, the appearance of the medial occipitotemporal and posterior parahippocampal gyri on the left was abnormal, with slight cortical thickening and indistinct gray-white matter differentiation, such that a focal cortical dysplasia was suspected. Intracranial electroencephalography suggested focal seizure onset from the suspect area, but the hypothalamic hamartoma was not explored. Resection of this area of cortex confirmed cortical dysplasia; the microscopic findings included atypical neurons with enlarged nuclei, but the patient’s seizures continued.
Discussion
MR Signal Intensity and Metabolite Profile
Increased T2-weighted signal intensity relative to gray matter, lack of contrast enhancement, and stable lesion size are the most frequently reported MR imaging features of hypothalamic hamartomas (28), and these were confirmed by the present study. Unlike most studies, however, we found slightly decreased T1-weighted signal intensity relative to gray matter in almost three-quarters of our epileptic patients. Previous reports of hypothalamic hamartoma isointensity with gray matter used conventional spin-echo imaging (23, 29–34), in some cases only after IV administration of contrast material (22). Few examples of hypothalamic hamartoma hypointensity shown by conventional spin-echo imaging are found in the literature (35, 36). Our observations may be explained by greater signal intensity contrast among gray matter, white matter, and abnormal tissue obtained with inversion-prepared gradient-echo sequences (such as used in the present study), compared with conventional spin-echo sequences (37). The clinical relevance of T1-weighted and, in particular, T2-weighted contrast between hypothalamic hamartomas and the adjacent hypothalamus is readily apparent in the context of epilepsy surgery, with which the ability to distinguish between hamartoma and hypothalamus is of paramount importance in planning the surgical approach and during the procedure (8, 38). We think that the T2-weighted fast spin-echo sequences of our presurgical imaging protocol provided such contrast (see Figs 5 and 6).
The neurons of hypothalamic hamartomas resemble those of the normal hypothalamus microscopically (39), and the histopathologic findings in this series were in keeping with previous descriptions. Suggestions that epileptogenic hypothalamic hamartomas are distinguished by the presence of dysplastic neurons (12) and balloon cells (6) seem to have resulted from misinterpretation of a previous report (2). However, considerable variation in the relative contribution of neuronal and glial elements can be found, with some authors describing glial cell numbers similar to those seen in normal gray matter (40) and others reporting varying degrees of gliosis, from mild and moderate (33) to dense (41). Different cellular and connective tissue composition of the hypothalamic hamartomas compared with the normal hypothalamus is likely to account for the different signal intensity characteristics we observed in the present series.
Proton MR spectroscopy indicated that the hypothalamic hamartomas in our patients have lower total N-acetylaspartate content and higher myoinositol concentration than either normal thalamic gray matter or frontal lobe tissue composed of both gray and white matter in our normal volunteers. Low N-acetylaspartate concentration is generally interpreted as either neuronal loss or dysfunction (42). A previous qualitative spectroscopic study of five patients with hypothalamic hamartomas found a lower N-acetylaspartate:creatine ratio in the hamartomas as compared with the hypothalamus of normal volunteers (43). The authors suggested that their findings implied neuronal dysfunction within the hypothalamic hamartomas; however, they acknowledged that there is no analogous region in the normal brain that can serve as a control. Our findings could equally be interpreted as reduced neuronal density in the hypothalamic hamartomas compared with other brain tissues. Myoinositol is specific to glial cells, and an increase is most often interpreted as reflecting gliosis (42). The finding of increased hypothalamic hamartoma myoinositol is consistent with an increase in glial tissue compared with normal brain and may also explain the high T2-weighted signal intensity in comparison with normal gray matter that we observed in 93% of our cases. A quantitative study of glial cell numbers in excised hypothalamic hamartoma tissue correlated with MR signal intensity properties (metabolite spectra or T2-relaxometry) could resolve this issue.
Topography and Epileptogenesis
Hypothalamic hamartomas have been classified on the basis of the breadth of attachment to the tuber cinereum—sessile versus pedunculated (33), the presence of more than minimal distortion of the outline of the third ventricle—intrahypothalamic versus parahypothalamic (22), or by using a combination of size, breadth of attachment, distortion of the hypothalamus proper and location of attachment—types Ia, Ib, IIa, and IIb presented by Valdueza et al (21). Some agreement exists among studies that hypothalamic hamartomas associated with epilepsy have sessile attachment to the hypothalamus and displace normal hypothalamic structures, whereas those associated with precocious puberty alone may be pedunculated. Some would classify hypothalamic hamartomas associated with epilepsy according to their preferred microsurgical treatment approach (10).
Our findings largely confirm those of previous small studies in relation to hypothalamic hamartoma topography. The predominantly intraventricular hypothalamic hamartoma is a sessile lesion that corresponds to the intrahypothalamic variety reported by Arita et al (22) and to type IIa presented by Valdueza et al (21); these are the smallest epileptogenic hypothalamic hamartomas. We found that some small hypothalamic hamartomas sat atop the mammillary bodies and did not involve the tuber cinereum (Fig 1A). Patients with such lesions did not develop precocious puberty, which was associated with larger hamartoma size and contact by the hamartoma with the pituitary stalk (Table 1). Because larger hamartoma size in this series was associated with involvement of more anterior hypothalamic regions, it is not possible to say which of the identified factors is key. Small interpeduncular hamartomas are typical for patients with precocious puberty alone, and it has often been stated that among patients with hypothalamic hamartomas and precocious puberty, larger hamartoma size is associated with epilepsy (1, 44, 45). Precocious puberty in hypothalamic hamartomas may be due to autonomous luteinizing hormone-releasing hormone secretion within the hamartoma (46) or induction of hypothalamic pubertal neurendocrine function by secretion of transforming growth factor α (47). It is possible that the location of a hamartoma in relation to the tuber cinereum (predominantly intraventricular versus predominantly interpeduncular) reflects its cellular composition and neuroendocrine potential; large hamartoma size may merely indicate a greater likelihood of combined endocrine and neurologic clinical features.
Arita et al (22) suggested that only the intrahypothalamic type of hypothalamic hamartoma had ample neuronal networks through which epileptogenic neuronal activity could propagate. Notwithstanding the considerable variation in size and differences in degree and direction of extension of epileptogenic hypothalamic hamartomas in this series, the only common feature of all hamartomas in our patients with epilepsy was a connection to one or both mammillary bodies. Particularly well seen in thin section axial view T2-weighted images is the propensity of epileptogenic hypothalamic hamartomas to displace adjacent hypothalamic nuclei and the course of myelinated hypothalamic fiber tracts. The mammillary bodies and their projections, including the anterior thalamic nuclei, are involved in the generation of generalized convulsive seizures elicited by systemic pentylenetetrazol (48). Interruption of both mammillothalamic tracts prevents pentylenetetrazol seizures in experimental animals (19), as does high frequency electrical stimulation of mammillary nuclei (49). In a report of one patient with hypothalamic hamartoma and intractable epilepsy, a stereotactic lesion of one mammillothalamic tract resulted in cessation of generalized seizures whereas partial seizures persisted (9). Although the hypothalamus has many neocortical, limbic, subcortical and brain stem connections (26, 50), the intimate relation of hypothalamic hamartomas to the mammillary bodies and the disposition of the intrahypothalamic component of the hamartoma, encircled by the postcommissural fornix and mammillothalamic tract, suggest a role for these pathways in seizure propagation and perhaps in the progressive epileptogenesis of the syndrome (20).
Associated Brain Malformations
Malformations of cortical development have been described in patients with Pallister-Hall syndrome, in which hypothalamic hamartoma and central polydactyly may be associated with dysplastic nails, pituitary hypoplasia or dysfunction, bifid epiglottis, imperforate anus, and malformations affecting other systems (51). Few examples of nonsyndromal hypothalamic hamartomas with malformations of cortical development are reported, but the additional cerebral malformations in those cases appear similar to those described in cases of Pallister-Hall syndrome (52). Lange-Cosack (53) reported autopsy findings in one patient that included right frontal microgyria and periventricular heterotopia. Schmidt et al (54) described two patients with autopsy findings of right hemisphere microgyria and periventricular heterotopia in one case and heterotopic gray matter with partial agenesis of the corpus callosum in the other. Diebler and Ponsot (1) reported one patient with macrocephaly and evidence of extensive brain malformation shown by CT, including multiple noncommunicating cysts, agenesis of the corpus callosum, and cystic dilation of the cisterna magna. A recent report of one case noted an association with agenesis of the corpus callosum, heterotopic gray matter, and Dandy-Walker complex (55). In the present series, no patients with Pallister-Hall syndrome were included and associated malformations of cortical development were rare, occurring in only two referred patients with refractory epilepsy. We are aware of only three other such patients, one with bilateral schizencephaly (R. Kuzniecky, personal communication, August 2002), one with agenesis of the corpus callosum, culpocephaly, and periventricular heterotopia (J. Zempel, personal communication, February 2003), and one with extensive left mesial frontoparietal pachygyria and heterotopia (P. Shillito, personal communication, June 2003).
The importance of an association between hypothalamic hamartomas and malformations of cortical development is threefold. First, the rarity with which the two apparently combine makes it unlikely that earlier hypotheses concerning the role of additional brain malformations in the genesis of the epileptic and cognitive-behavioral features of the syndrome (1, 23) are correct. Second, the investigation of patients with hypothalamic hamartomas and intractable epilepsy should include MR imaging and interpretation of a sufficient standard to detect associated malformations of cortical development; in the rare event that the two are found together, further investigation may be required to determine their relative contribution to the patient’s neurologic presentation. Third, the pattern of combined cortical and subcortical malformation observed in both syndromal and nonsyndromal hypothalamic hamartomas may indicate a common mechanism for or antecedent to their development. Although mutations in the transcription factor gene GLI3 cause Pallister-Hall syndrome (56, 57), the relationship between GLI3 and nonsyndromal hypothalamic hamartomas is as yet undefined.
Four patients (6%) in the present series had arachnoid cysts. Two other patients had cysts located entirely within the hamartoma, an observation noted previously in cases of large hypothalamic hamartoma (58). Of interest is the spatial relationship between the cysts and the hypothalamic hamartoma. Previous reports have noted that the arachnoid cyst lies adjacent to the hamartoma, which may itself have a cystic component (59–61) or be compressed by the arachnoid cyst (62). The findings for our patients suggest a spectrum of associated cystic abnormalities, including intrahamartomatous cysts (Fig 3), arachnoid cysts that extend within the hamartoma, and arachnoid cysts that are adjacent to or remote from the hypothalamic hamartomas (Fig 9). The cause of congenital intracranial arachnoid cysts is not known, but abnormal splitting of the developing arachnoid membranes has been proposed to relate to focal maldevelopment of the meninges during embryonic folding of the neural tube (63) and in proximity to the normal arachnoid cisterns (64), accounting for both their histopathologic features and characteristic locations. Genetic factors have been implicated in some cases, including a report of familial occurrence (65), monozygous twins with different temporal lobe malformations (schizencephaly in one and arachnoid cyst in the other) (66), and association of bitemporal arachnoid cysts with glutaric aciduria type 1 (67) and neurofibromatosis (68). Although there are no population-based studies of the prevalence of arachnoid cysts, their incidence has been estimated at five per 1000 necropsies (69); this frequency is an order lower than that found in the present series, making a chance association unlikely. Whether the co-occurrence of hypothalamic hamartomas and arachnoid cysts is a result of mechanical influence exerted by the hypothalamic hamartomas during subarachnoid space formation (61) or some other mechanism is speculative.

Arachnoid cysts associated with hypothalamic hamartomas.
A, T1-weighted coronal view MR image shows a large suprasellar and temporal fossa arachnoid cyst, hypoplasia and compression of the right temporal lobe (bloom artifact due to previous cyst-peritoneal shunt insertion), and cavum septum pellucidum (arrow).
B, T1-weighted coronal view MR image shows a suprasellar arachnoid cyst extending within the hamartoma.
C, T2-weighted axial view MR image shows an anterior temporal arachnoid cyst (arrowhead) remote from the hamartoma (not shown).

Malformations of cortical development associated with hypothalamic hamartomas in two patients.
A, T2-weighted axial and coronal view MR images in the first case show periventricular nodular heterotopia and focal dilation of the left lateral ventricle, plus overlying calcarine and temporal-parietal-occipital junction cortical thickening and abnormal gyral configuration (white arrows). The hypothalamic hamartoma is seen in the axial view image (arrowhead).
B, Inversion recovery coronal view image obtained at the level of the posterior hippocampus in the second case shows an atypical configuration of the medial occipitotemporal and posterior parahippocampal gyri (black arrow), with slight cortical thickening and indistinct gray-white boundary.
Temporal Lobe Findings
A somewhat surprising finding, considering the refractory and longstanding nature of the epilepsy that occurs in most patients with hypothalamic hamartoma, is the absence of a single case of hippocampal sclerosis. Even in patients who, because of pseudolocalizing clinical and EEG information, had undergone temporal lobe resections in an attempt to control complex partial seizures, the mesial temporal structures were normal. The finding may be explained by the absence of a history of convulsive status epilepticus in these patients, consistent with the view that in temporal lobe epilepsy at least, hippocampal sclerosis results from an initial precipitating injury rather than as a consequence of repeated seizures (70, 71).
Anterior temporal lobe signal intensity abnormalities (increased T2-weighted white matter signal intensity with loss of gray-white matter differentiation) comprised the most frequent associated finding, occurring in 16% of patients. This proportion, however, is considerably less than that reported in patients with temporal lobe epilepsy; approximately two-thirds of those patients have these appearances, usually in association with hippocampal sclerosis (72). Debate surrounds the significance of these anterior temporal abnormalities. Some authors assert that they are a developmental malformation associated with an increase in ectopic white matter neurons (73, 74), whereas others consider that the changes reflect abnormal myelination or demyelination in the temporal lobe (72, 75), an hypothesis supported by the finding of associated reduced temporopolar white matter volume (76) and by a recent study of children with hippocampal sclerosis, in whom anterior temporal signal intensity changes were associated with a very early onset of seizures (77). If the latter hypothesis is correct, anterior temporal lobe changes observed in patients with hypothalamic hamartomas could be a result of seizure propagation from the hypothalamic hamartomas to the temporal lobe. Some evidence for the involvement of the anterior temporal lobe in seizures associated with hypothalamic hamartomas comes from a series of eight patients studied with intracranial EEG, seven of whom had apparent (although falsely localizing) unilateral onset of seizures from the anterior temporal lobe (78). That the unilateral anterior temporal abnormalities in our patients were always ipsilateral to the side of predominant hamartoma attachment to the hypothalamus also suggests some influence of the hypothalamic hamartomas over the temporal changes observed. Further study of seizure origin and spread in patients with hypothalamic hamartoma may clarify this relationship.
Conclusion
Epileptogenic hypothalamic hamartomas are malformations of the mammillary region of the hypothalamus and are invariably attached to one or both mammillary bodies. MR signal intensity characteristics and spectroscopic findings suggest reduced neuronal density and relative gliosis and help to distinguish the hamartoma from the surrounding deep gray matter. The intrahypothalamic component lies in the wall of the third ventricle between the postcommissural fornix anteriorly, the mammillothalamic tract posteriorly, and the mammillary body inferiorly, suggesting a role for these structures in seizure propagation and epileptogenesis. Associated malformations of cortical development are rare, occurring in only 3% of patients in this series, and are unlikely to contribute to the typical clinical syndrome. An association with arachnoid cysts suggests either a common antecedent or causal relationship. Hippocampal sclerosis is not associated with hypothalamic hamartoma, but anterior temporal white matter changes seen in a small proportion may reflect ictal involvement of the temporal lobe during postnatal brain development.
Footnotes
This work was presented at the Australian Association of Neurologists Annual Scientific Meeting, Sydney, 2003.
This work was supported by scholarships from the National Health and Medical Research Council (to J.L.F.), the Murdoch Children’s Research Institute, and the University of Melbourne Faculty of Medicine, Dentistry and Health Sciences.
References
- Received May 1, 2003.
- Accepted after revision August 26, 2003.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Outcomes following surgical interventions for hypothalamic hamartomas: protocol for a systematic review and individual patient data meta-analysis
- The Mammillary Bodies: A Review of Causes of Injury in Infants and Children
- Hypothalamic Hamartomas: Evolving Understanding and Management
- The Frequency and Extent of Mammillary Body Atrophy Associated with Surgical Removal of a Colloid Cyst
- The diagnosis and management of parasellar tumours of the pituitary
- One-year-old male with accelerated growth and development