
Abstract
Summary: We report a case of a supratentorial primitive neuroectodermal tumor (PNET) that occurred 12 years after cranial irradiation for a grade II astrocytoma. Neuroimaging was unable to distinguish between a recurrence of the original neoplasm and the development of a new, distinct entity. Pathologic review assisted by immunohistochemical staining, however, revealed a high-grade PNET. Although rare, PNET needs to be included in the differential diagnoses for previously irradiated patients, who develop recurrent brain tumors in the presence of uncharacteristic imaging features.
Tumors of the CNS are infrequent sequelae of cranial irradiation. These radiation-associated tumors may manifest as gliomas, meningiomas, schwannomas, sarcomas, and primitive neuroectodermal tumors (PNETs; 1–3). We describe a supratentorial PNET arising in a patient 12 years after surgery and cranial irradiation for a low-grade astrocytoma.
Case Report
A 27-year-old woman presented with a grand mal seizure. Her workup revealed a right frontal brain mass for which she underwent surgical resection. The pathology department reported a grade II astrocytoma; unfortunately, slides from this surgery are no longer available for review. Postoperatively, the patient received 4500 cGy to the whole brain with a tumor bed boost to a total dose of 5040 cGy at a local community hospital. She was followed closely for 5 years, during which time there was no evidence of recurrence.
She did well until 12 years after completion of irradiation, when she developed sinus pressure and fatigue. Her symptoms progressed, and she presented to the emergency department complaining of nausea and vomiting. CT (not shown) revealed a large right frontal lobe intraaxial mass, and MR imaging revealed a well-circumscribed, heterogeneously enhancing mass measuring 5.1 × 6.4 cm, with surrounding edema, mass effect, and midline shift (Fig 1A and B). The tumor was slightly hypointense relative to gray matter on noncontrast T1-weighted images and slightly hyperintense on T2-weighted images (not shown). On diffusion-weighted imaging, the lesion demonstrated markedly increased signal intensity (not shown). Central necrosis was absent. The patient underwent a right frontal craniotomy with microdissection and resection of the tumor (Fig 1C). Histopathologic review of the specimen revealed an embryonal tumor composed of poorly differentiated neuroepithelial cells with pseudopalisading necrosis and endothelial proliferation and a high mitotic index on Ki-67 immunostaining (Fig 2A). Immunohistochemical staining showed scattered tumor cells staining positively for GFAP and many cells with positive cytoplasmicstaining for synaptophysin and negative staining for neurofilament, indicating divergent neuroblastic and glial differentiation. (Fig 2B and C). The tumor was classified as a World Health Organization grade IV supratentorial PNET.

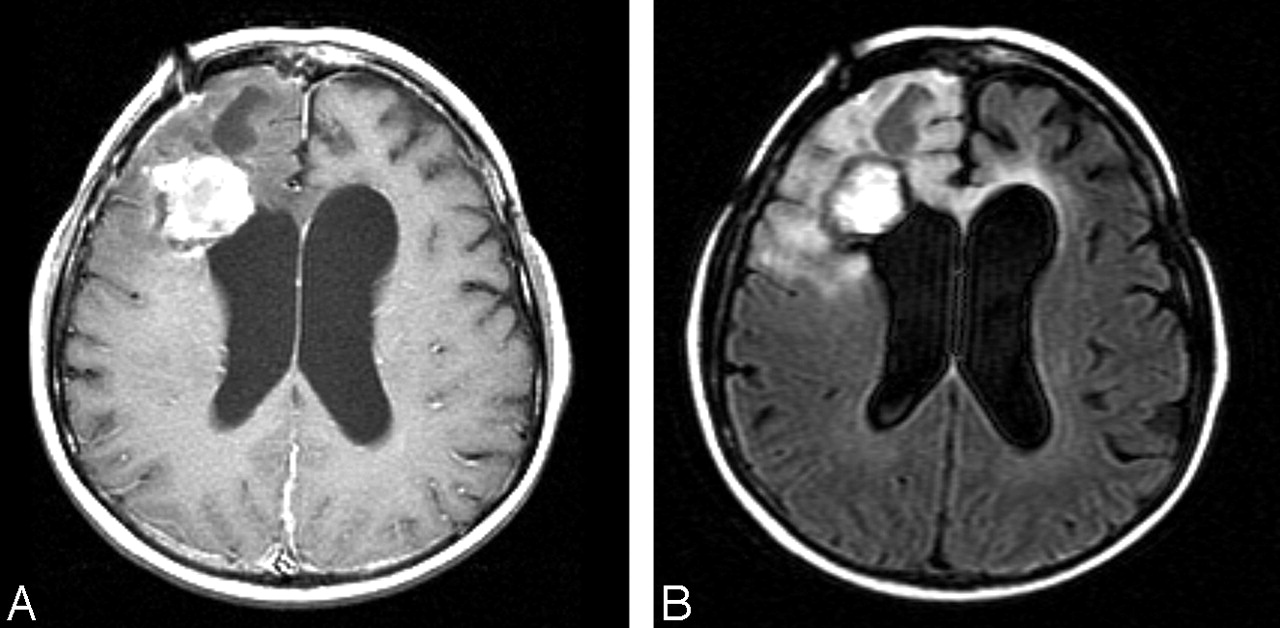
MR imaging shows a 5.1 × 6.4 cm heterogeneous mass in the right frontal lobe with areas of necrosis and surrounding edema.
A, Axial T1-weighted postgadolinium image (500/8/2; TR/TE/NEX).
B, Axial FLAIR image (8002/158/1; TI, 2000).
C, Axial FLAIR image (8002/158/1; TI, 2000) immediately after surgical resection.
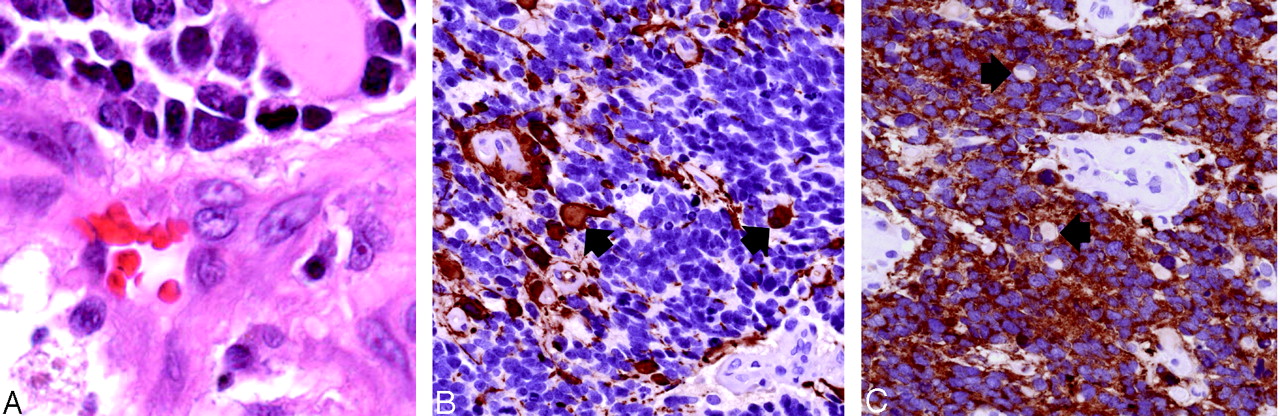
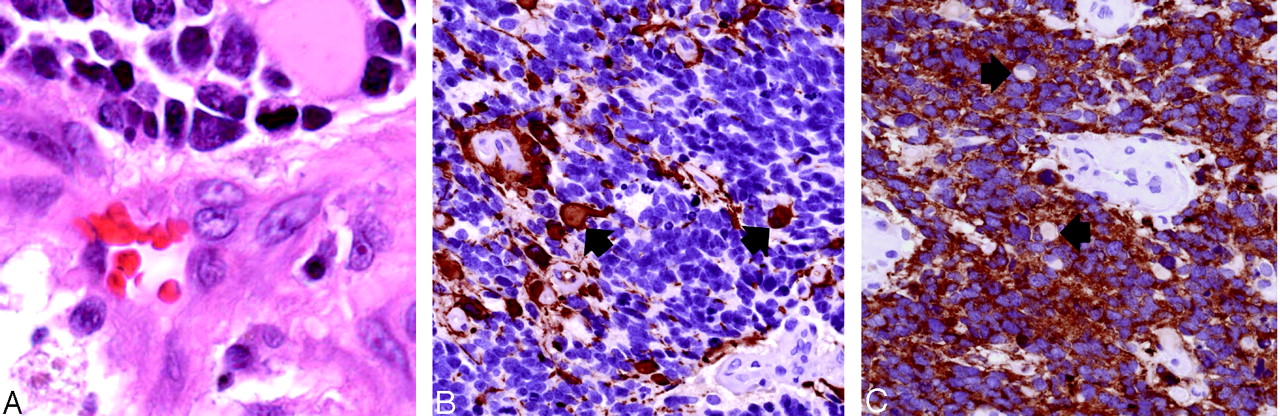
Histopathologic findings (original magnification ×400).
A, Hematoxylin-eosin stained sections show sheets of undifferentiated tumor with frequent apoptotic figures and endothelial proliferation. Malignant gemistocytes indicate astrocytic differentiation (arrows).
B, Immunostaining with anti-GFAP antibody shows positive staining of fibrillary tumor cell processes as well as of the rounded cell bodies of malignant gemistocytic astrocytes (arrows).
C, Scytplasmic staining of many tumor cells, indicating neuroblastic differentiation. Cells showing absent cytoplasmic staining (arrows) correspond to cells with astrocytic differentiation.
The patient’s postoperative recovery was complicated by multiple pulmonary emboli; however, she recovered well. Postoperative radiation therapy was not an option for this patient because of her previous full-dose of radiation to the same area. The patient was later treated with chemotherapy, and follow up imaging 4 months after surgery revealed a stable appearance of the mass (Fig 3A and B).
Follow-up MR imaging 4 months after resection shows a stable appearance of her right frontal tumor with decreased edema and mass effect.
A, Axial T1-weighted postgadolinium image (400/8/2).
B, Axial FLAIR image (8002/158/1; T1, 2000).
Discussion
The development of brain tumors in previously irradiated sites is a rare complication of cranial irradiation (1, 2). Other possible risk factors for these secondary CNS tumors include younger age at irradiation, genetic predisposition to cancer, and genetic polymorphisms in certain metabolizing enzymes, such as thiopurine s-methyl-transferase (2). The most commonly reported histologic types of these tumors are the high-grade gliomas, including glioblastomas and malignant astrocytomas (1). Nevertheless, cases of meningiomas, schwannomas, sarcomas, and PNETs arising in the CNS after cranial irradiation have also been described (1–5). Secondary PNETs have been reported in 11 patients at a mean of 8.5 years following a relatively low-dose prophylactic craniospinal irradiation (18–24 Gy) and intrathecal methotrexate for leukemia and lymphoma (3). In addition, a supratentorial secondary PNET was reported to occur in a patient with unilateral sporadic retinoblastoma 5 years after surgery, low-dose cranial irradiation (27.6 Gy), and intrathecal methotrexate (4). In these cases, the relative contribution of low-dose irradiation versus intrathecal methotrexate to the development of PNET is not clear.
Hader et al (4) recently reported four cases of PNETs developing in the CNS after 50–55 Gy of irradiation alone for low-grade intracranial neoplasms. Our case is similar to those reported by Hader et al and represents the fifth incidence of PNET that is directly associated with high dose of irradiation.
PNETs describe a group of histologically similar neoplasms composed of embryonal small cells that may be undifferentiated or may have varying degrees of neuronal, glial, or mesenchymal differentiation. PNETs of the CNS include medulloblastomas, neuroblastomas, pineoblastomas, and ependymoblastomas. The exact mechanism of the development of PNETs after cranial irradiation is unknown, although it has been speculated to involve the persistence of a population of undifferentiated neuroepithelial cells in the CNS, as well as genetic mutations involving oncogenes such as K-ras (3, 5).
Development of a distinct tumor at the site of prior irradiation can be difficult to distinguish from recurrent primary tumor or radiation necrosis. In this case, the 12-year time interval between the initial treatment and the appearance of a new lesion argues against the latter two. The imaging characteristics also would be unusual for astrocytoma or radiation-induced injury. The relatively low signal intensity on T2-weighted images and high signal intensity on diffusion-weighted images imply the presence of dense cellularity and high nuclear-to-cytoplasmic ratio, which would not be expected with low-grade astrocytoma or radiation necrosis and that can be seen in PNETs. In addition, the lack of central necrosis argues against radiation necrosis or dedifferentiation of the original tumor into a more high-grade glial neoplasm. MR spectroscopy or positron emission tomography could be used to distinguish between these entities, but in this case, the diagnosis was made histologically, because the patient required surgery for relief of mass effect.
The reported survival time in patients with radiation-associated PNETs of the CNS is rather short. The four patients described by Hader et al had a mean survival of 12 months after diagnosis; however, only two of these patients received chemotherapy (5). This short survival time may be attributed to the aggressive nature of tumors that develop after irradiation, as well as the limited treatment options available to patients who have previously received full-dose irradiation to the tumor area. Our patient remains stable both clinically and radiologically after two cycles of combination chemotherapy, including cytoxan, cisplatin, etoposide, and vincristine.
- Received December 10, 2003.
- Accepted after revision March 16, 2004.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.