
Abstract
Summary: Hypothalamic hamartoma is a rare congenital lesion. We present the case of a 7-year-old girl who suffered from precocious puberty, the cause of which was diagnosed by using MR imaging and CT as pedunculated hypothalamic hamartoma associated with a large craniopharyngeal canal and sellar spine mimicking pituitary duplication.
Hypothalamic hamartoma is a rare congenital, nonneoplastic heterotopia and is an important cause of central precocious puberty. The large craniopharyngeal canal is a rare congenital skull base defect. The association of hypothalamic hamartoma and large craniopharyngeal canal without duplication of the pituitary gland has been described. We report such a case with associated sellar spine mimicking pituitary duplication. The diagnosis was based on CT and MR imaging findings.
Case Report
A 7-year-old girl presented with growth of breasts and beginning of menarche. Physical examination revealed mild enlargement of breasts, pubic hair, and prominent labia. The neurologic examination was within normal limits, and no anomaly was detected on physical examination. Serum levels of sexual hormones such as LH, FSH, and E2 reached the levels of puberty or adulthood. Bone age was consistent with that of a girl 10 years old. Abdominal ultrasonography revealed that the uterus and ovaries had peripubertal morphology.
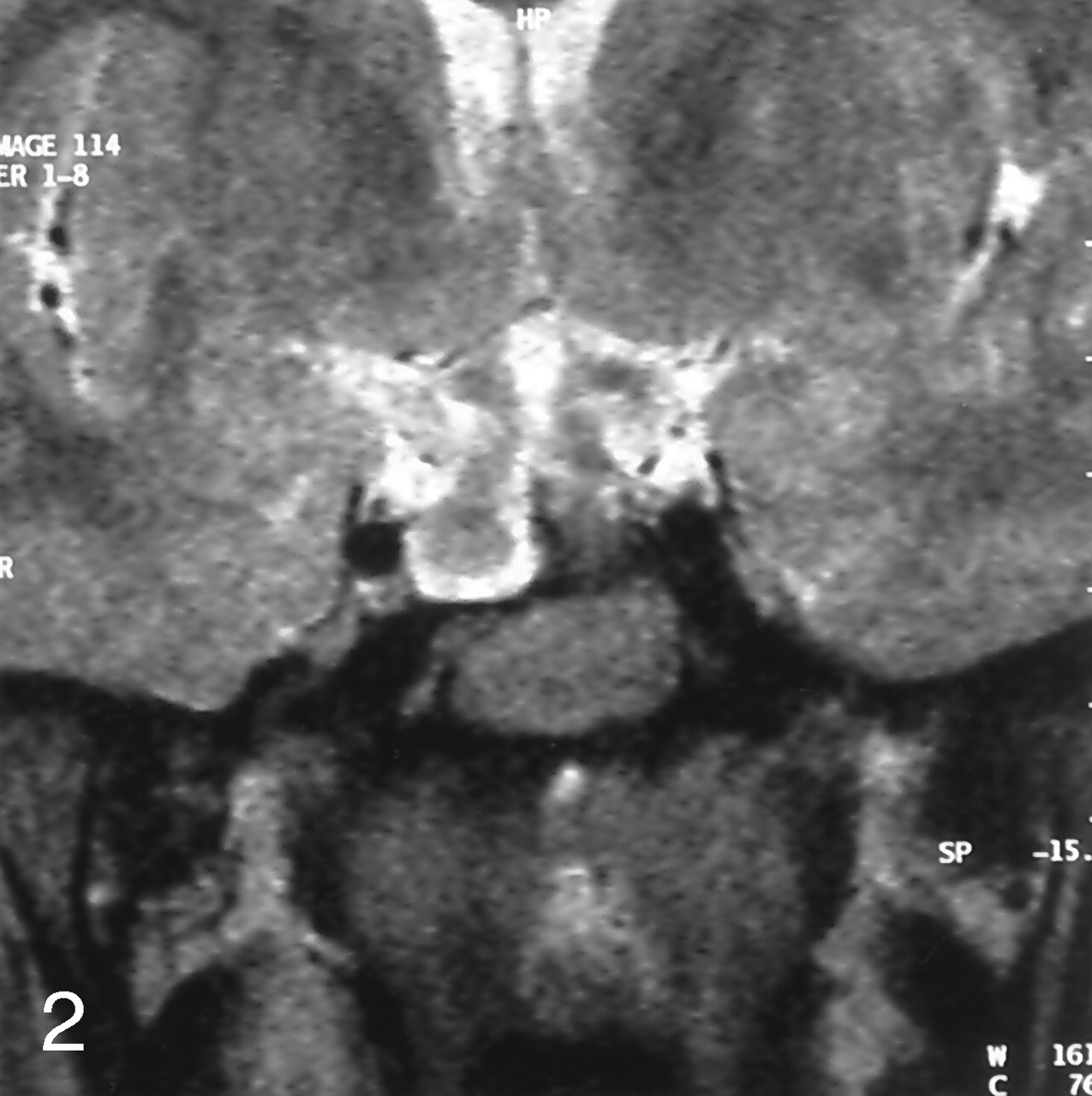
MR imaging of the brain showed a sellar spine dividing the sellar cavity into two halves, a pedunculated hypothalamic mass arising from the tuber cinereum that was isointense to brain parenchyma on both T1- and T2-weighted images, 1 cm in diameter, extending into suprasellar cistern and right half of the sella turcica. The pituitary gland was located in the left half of sella turcica and was displaced minimally downward from the midline defect. These precontrast series resembled duplication of the pituitary gland, associated with midline bone defect and sellar spine. After contrast medium administration, homogeneous enhancement was seen in the pituitary gland and stalk, located in the left side of the sella turcica, whereas the lesions on the right side and its peduncle were not enhanced. With these findings, we concluded that association of the pedunculated hamartoma and the midline defect resulted from the left-sided location of the normal pituitary gland and infundibular stalk, mimicking duplication of the pituitary gland (Figs 1 and 2).

Coronal spin-echo T1-weighted precontrast image (A) shows the association of hypothalamic hamartoma and pituitary gland mimicking the duplicated pituitary gland, and postcontrast image (B) demonstrate difference of contrast enhancement pattern. Note the homogeneous contrast enhancement of pituitary gland and lack of contrast enhancement in the hypothalamic hamartoma.

Coronal turbo spin-echo T2-weighted image shows the hypothalamic hamartoma arising from the tuber cinereum, sellar spine, and lateral displacement of pituitary gland.
Coronal CT of the skull base was performed to better delineate the bone structure. A bone defect, consistent with large craniopharyngeal canal extending from the floor of the sella turcica to the nasopharynx and sellar spine was apparent on CT images. The sellar cavity was divided into two spaces by the sellar spine: the right half of the sellar cavity contained the hamartoma, and the left half of the cavity contained the pituitary gland. Downward displacement of pituitary gland through the midline bone defect was also seen. Pituitary gland attenuation was homogeneous, and a mass lesion located in the right side of the sella was isoattenuated to brain parenchyma (Fig 3). The final diagnosis was made, on the basis of both MR imaging and CT findings, as a midline bone defect (large craniopharyngeal canal) and sellar spine resulting from displacement of the pituitary gland associated with a pedunculated hamartoma, mimicking duplication of pituitary gland. Repeated physical examination did not reveal any craniofacial abnormalities.
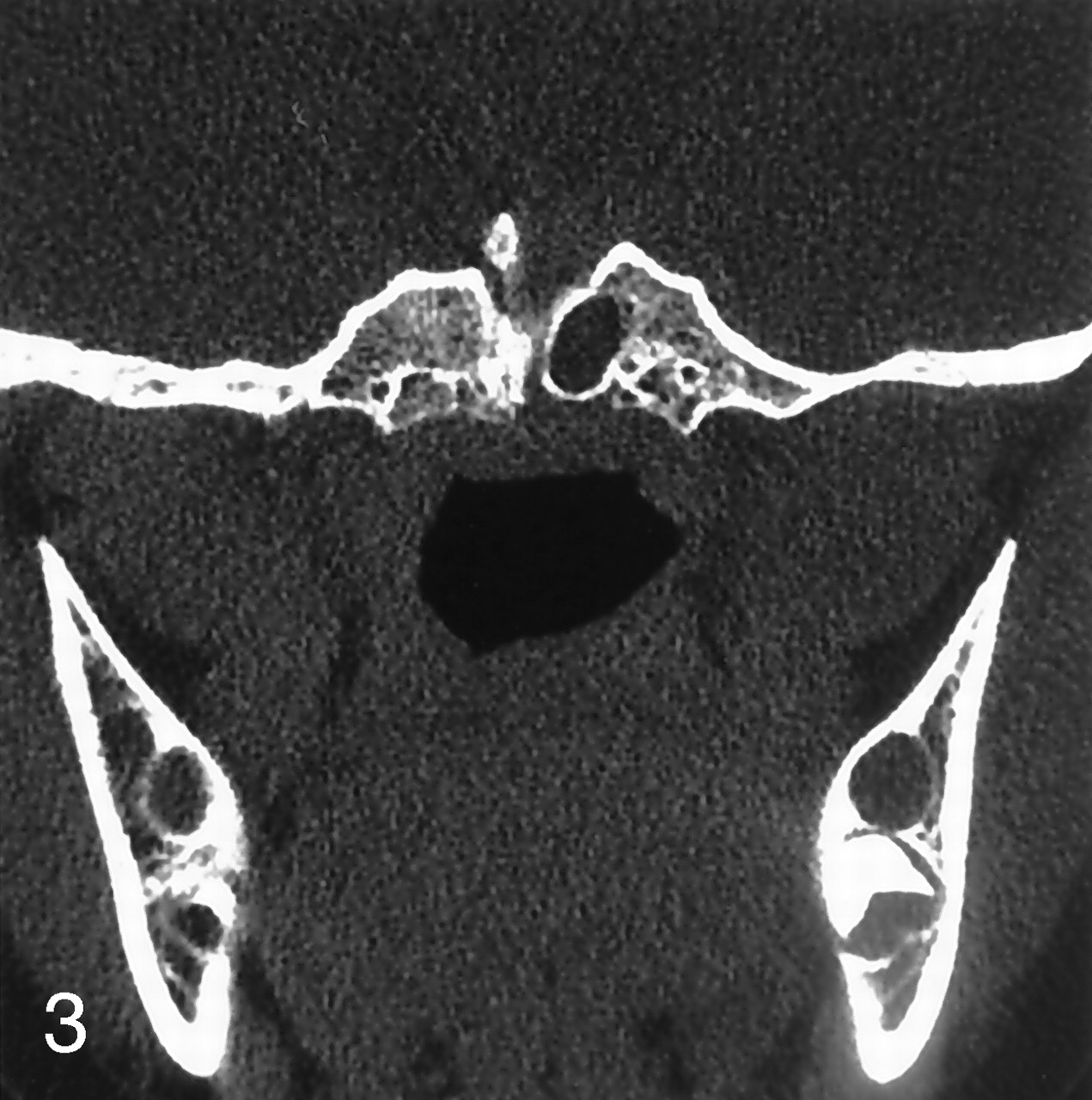
Coronal CT scan delineates the large craniopharyngeal canal and associated sellar spine.
Treatment requires monthly administration of medication with a gonadotropin-releasing hormone (GnRH) analog until puberty. The patient was started on a GnRH analog, after which there was regression of the pubertal development and suppression of hormone levels. Follow-up MR imaging, however, did not show any difference of the hamartoma at the 6th month of medication.
Discussion
Hamartomas of the tuber cinereum are rare congenital nonneoplastic heterotopias. Most of the neurons in a hamartoma are similar to normal hypothalamic neurons (1). Diagnosis is based on the characteristic location, isointensity to normal brain, lack of contrast enhancement, and absence of change in size and morphology of the mass at the follow-up. Differential diagnosis of hypothalamic hamartoma may include craniopharyngioma and optic-hypothalamic gliomas. These tumors have heterogeneous signal and often show contrast enhancement, whereas hypothalamic hamartomas do not show contrast enhancement (2). Boyko et al (3) classified hypothalamic hamartomas into two types: the pedunculated type is more likely to be associated with precocious puberty, and the sessile type is often associated with gelastic seizures. Hypothalamic hamartomas <1 cm usually present with precocious puberty, whereas lesions >1 cm usually present with gelastic seizures. The most common tumoral cause of central precocious puberty is hypothalamic hamartoma, the incidence ranging from 14–58% (4).
Duplication of the pituitary gland is an extremely rare malformation. MR imaging reveals two pituitary glands and infundibular stalks. The hypothalamic hamartoma is the most common abnormality associated with duplication of the pituitary gland, and it occurs most commonly in association with midline craniofacial anomalies such as craniopharyngeal (persistent hypophyseal) canal, large craniopharyngeal (transsphenoidal) canal, median cleft syndrome, and basal cephaloceles (4–6).
The spectrum of congenital abnormalities affecting the skull base ranges from the craniopharyngeal canal, which connects the pituitary fossa and nasopharynx, to large basal cephaloceles with craniofacial defects (7). The term “craniopharyngeal canal” is generally used to describe a small and vertical midline defect in the skull base that measures <1.5 mm in diameter. Currarino et al (8) provided other names to describe this skull base defect, including the large craniopharyngeal canal, which differs from the normal craniopharyngeal canal by virtue of greater size and association with special craniofacial anomalies. The midline bone defect detected in our patient was consistent large craniofacial canal, because it was >1.5 mm. The sellar spine is believed to be a congenital malformation with incomplete regression of the cephalic tip of the notochord. The persistence of a notochord rest within the fetal sella will form the ossified sellar spine.
The association of hypothalamic hamartoma and large craniopharyngeal canal has been reported (9). This is such a case of hypothalamic hamartoma associated with large craniopharyngeal canal without pituitary duplication, but it has a peculiar situation, mimicking pituitary duplication because of the location of the pedunculated hamartoma and pituitary gland. In our case, the association of the sellar spine with hypothalamic hamartoma and large craniopharyngeal canal was demonstrated. The diagnosis was based on contrast-enhanced MR imaging and CT findings of the basicranium.
The diagnosis of hypothalamic hamartoma, pituitary duplication, and associated craniofacial anomalies mainly depend on MR imaging, but CT better delineates bony defects. For this reason, a combination of MR imaging and CT is needed, as in the case presented here. Detailed physical examination is mandatory for detecting the presence of craniofacial anomalies.
The associated anomalies detected in our patient were consistent with notochordal anomalies. The time of induction to the teratogenic influence and extent of the notochordal abnormality may vary and account for the wide spectrum of associated abnormalities (5–7).
Treatment options for precocious puberty associated with hypothalamic hamartoma may be surgical or medical. Surgical excision of hamartoma in young children is recommended, whereas in children close to puberty, no surgical treatment is required. Medical treatment includes use of a long-acting GnRH analogue or antagonists until puberty (10, 11). Our patient did well with medical treatment, and regression of the pubertal development and suppression of hormone levels were observed in the follow-up.
Conclusion
The case of hypothalamic hamartoma associated with large craniopharyngeal canal presented here was diagnosed on the basis of CT and MR imaging findings. In this case, association of the sellar spine mimicked pituitary duplication. The use of both CT and MR imaging in this context is advocated.
References
- Received January 9, 2004.
- Accepted after revision March 29, 2004.
- Copyright © American Society of Neuroradiology









{kind=link}
{kind=link}
{kind=link}