
SUMMARY:
The so-called molar tooth sign is the radiologic hallmark of JSRD. Joubert syndrome is a rare, most often autosomal-recessive disorder with a characteristic malformation of the midhindbrain. We describe 3 patients with JSRD and the additional MR finding of tissue resembling heterotopia in the interpeduncular fossa, which in one patient was combined with a more extensive intramesencephalic heterotopia. Interpeduncular heterotopia has not been reported previously, either in the context of JSRD or as a separate entity. This new imaging feature enlarges the spectrum of brain stem abnormalities in JSRD. In view of the underlying ciliopathy, it seems likely that the interpeduncular heterotopia results from misdirected migration.
Abbreviations
- CNS
- central nervous system
- GE
- gradient-echo
- JSRD
- Joubert syndrome and related disorders
- T1WI
- T1-weighted imaging
- T2WI
- T2-weighted imaging
Joubert syndrome is a rare disorder (estimated prevalence 1/100 000) with a characteristic complex malformation of the midbrain-hindbrain, seen as the so-called molar tooth sign on axial imaging. This results from vermis hypoplasia, a deep interpeduncular fossa and thickened, elongated, abnormally horizontal superior cerebellar peduncles.1 Joubert syndrome is clinically characterized by cognitive impairment, hypotonia, and later evolving ataxia. An irregular breathing pattern in infancy and ocular apraxia are inconsistent, though typical findings. JSRD comprises all disorders with the molar tooth sign on imaging, namely, classical Joubert syndrome and further, related disorders with additional CNS, ocular, renal, hepatic, and/or skeletal manifestations.
The molar tooth sign is the mandatory criterion to diagnose JSRD. Although additional CNS changes may be present, interpeduncular heterotopia has not been described previously, either in conjunction with JSRD or as a separate entity. We present 3 patients with JSRD and a distinct interpeduncular heterotopia.
Cases
Patient 1 is the only child of unrelated Turkish parents. She presented with episodes of tachypnea and nystagmus in the neonatal period. Subsequently, global developmental delay and muscular hypotonia were noted. Ophthalmologic examination revealed diffuse retinal dystrophy. MR imaging at the age of 10 months confirmed the suspected diagnosis by demonstrating the diagnostic molar tooth sign. Molecular genetics revealed a homozygous mutation of CEP290, which is identified in approximately 50% of patients with the oculo-renal form of JSRD.2 There was no evidence of renal and/or hepatic involvement. At 26 months, her ability to maintain head and truncal control had significantly improved, and her respiration normalized. Neurologic examination revealed persisting profound muscular hypotonia and lack of spontaneous movements, horizontal nystagmus, and lack of fixation due to severely reduced visual acuity.
In addition to the characteristic molar tooth sign, MR imaging revealed a circumscribed, nodular structure within the interpeduncular fossa. This was isointense to gray matter and in direct contact with the mesencephalon at the level of the substantia nigra. Sagittal images confirmed the interpeduncular location without contact to hypothalamic structures (Fig 1). In addition, there was a small (approximately 2 mm) cyst within the right cerebral peduncle and asymmetry of the brain stem with a smaller left cerebral peduncle and pons. The cisternal segments of oculomotor, abducens, and trigeminal nerves were regularly identified on high-resolution, heavily T2-weighted images. No malformation of cortical development was detected in the cerebrum; the hemispheres were symmetric. Myelination at 10 months was delayed with incomplete myelination on T1-weighted images.
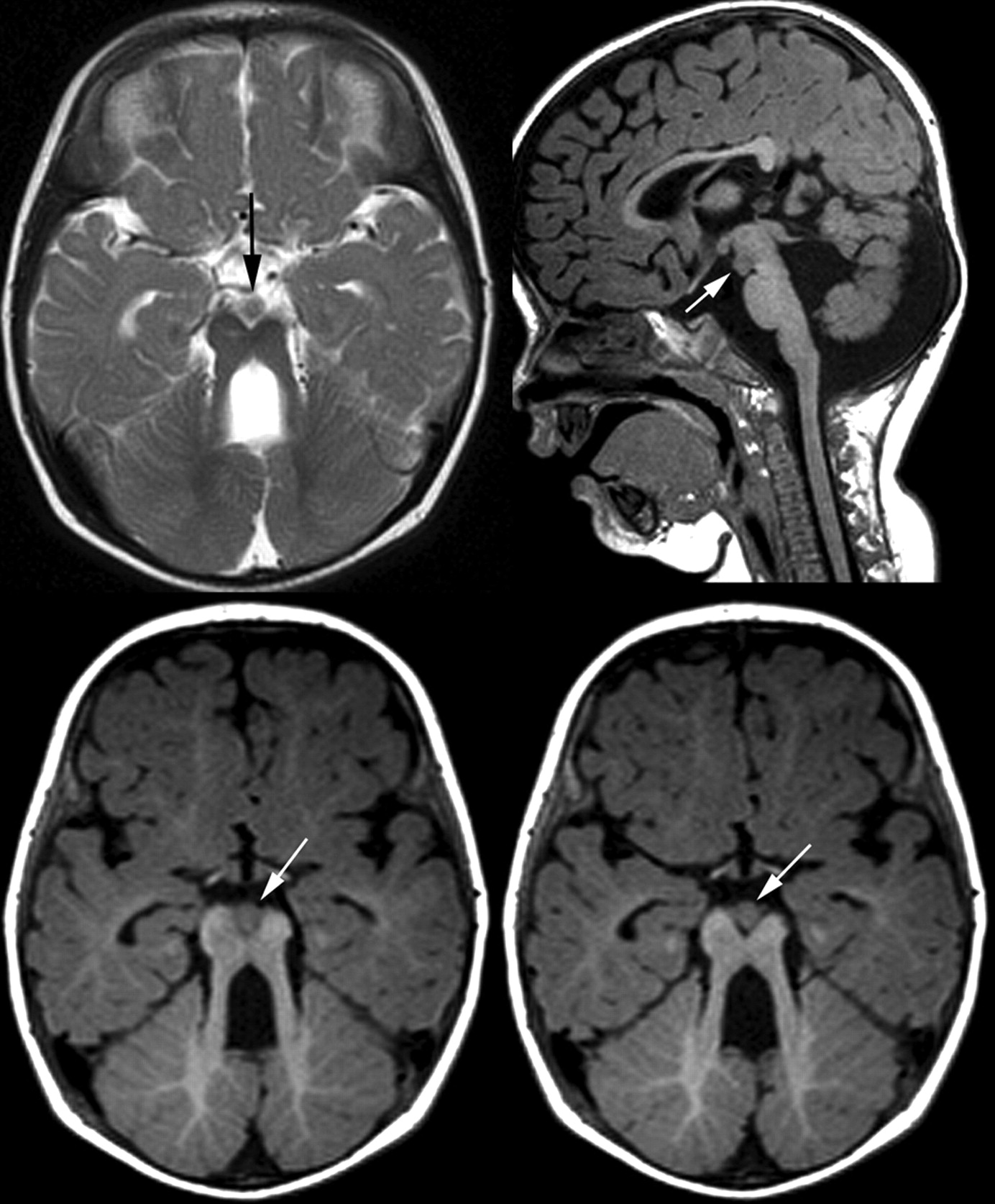
MR imaging of patient 1, aged 10 months. Heterotopic tissue within the interpeduncular fossa (arrows) appears continuous with the cerebral peduncles and without diencephalic connection (axial T2WI TSE, 5 mm; saggital T1WI TSE, 1 mm)
Patient 2 is the second child of nonconsanguineous French parents. His older brother was diagnosed with Joubert syndrome at the age of 1 year based on typical clinical and neuroimaging findings. The MR imaging of this older sibling revealed the characteristic molar tooth sign but no supratentorial abnormality, and in particular, no hypothalamic or premesencephalic lesion. In patient 2, Joubert syndrome was suspected in the neonatal period in view of respiratory abnormalities and facial phenotype. Molar tooth sign on MR imaging at 16 months confirmed the diagnosis. His development was delayed. Molecular testing did not yield a known mutation; there is however marked genetic heterogeneity, and mutations are currently identified in only approximately 50% of patients.
MR imaging demonstrated a nodular structure of gray matter isointensity within the interpeduncular fossa. Sagittal images confirmed the interpeduncular localization and absence of a connection with the hypothalamus (Fig 2). The cisternal segments of oculomotor, abducens, and trigeminal nerves were regularly identified on thin, heavily T2-weighted images. The vermis was hypoplastic with the typical cleft; the superior cerebellar peduncles were thickened, elongated, and horizontal, resulting in the diagnostic molar tooth sign. In this patient, there was no asymmetry of brain stem structures. Myelination was adequate for age.

MR imaging of patient 2, aged 16 months. Heterotopic tissue in the interpeduncular fossa (arrows) is continuous with the mesencephalon and clearly separate from the diencephalon (reconstruction of 3D T1WI GE, 1.2 mm and 3D T2WI GE, 1 mm).
Patient 3 is the only child of unrelated Swiss parents. At the age of 1 year, Joubert syndrome was diagnosed in view of his facial phenotype, respiratory abnormalities (episodes of apnea and tachypnea), and marked vermis hypoplasia on CT. His motor and language development was markedly delayed. When participating in an MR imaging research study at the age of 26 years (case 3 in that study)3 his neurologic examination revealed moderate bilateral ptosis, limitation of upward gaze, hoarse voice, moderate truncal ataxia, and limb dysmetria. There was no evidence of retinal and/or renal involvement. Genetic testing did not reveal a known mutation.
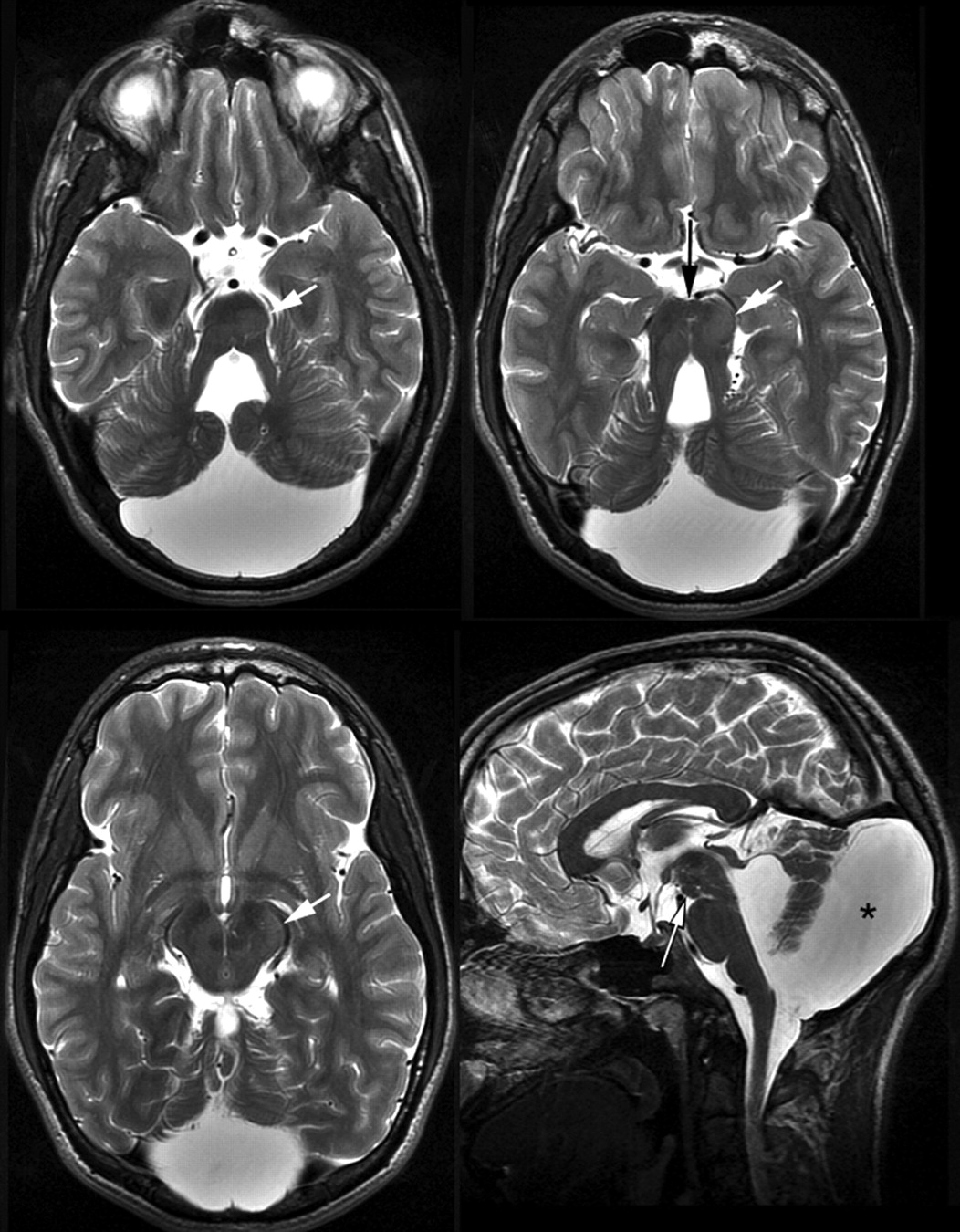
In patient 3, interpeduncular tissue was combined with an extensive heterotopia within mesencephalon and pons. The interpeduncular structure was nodular with direct lateral contact to the cerebral peduncles and no contact to diencephalic structures (Fig 3). The intraparenchymal heterotopia3 extended from the left dorsal paramedian basis pontis to the posterior limb of the left internal capsule. It enlarged the tegmentum and left cerebral peduncle and displaced the substantia nigra medially and dorsally. Marked asymmetry of the pontomesencephalic junction was at least partially due to the intraparenchymal heterotopia. The posterior fossa was enlarged with a large retrocerebellar cistern broadly communicating with the fourth ventricle. No malformation of cortical development was detected within the cerebrum; myelination was complete.

MR imaging of patient 3, aged 26 years. Interpeduncular tissue (long arrows) without connection with the hypothalamus (saggital and axial T2WI TSE, 3.5 and 3 mm). Note the intraparenchymal heterotopia (short arrows) and large posterior fossa and retrocerebellar cistern (asterisk).
Discussion
Nodular interpeduncular tissue with a signal intensity of normal parenchyma and direct contact to the cerebral peduncles was the distinct and previously unreported imaging finding in our patients, who otherwise presented with characteristic imaging and clinical findings of JSRD. We decided to use the term interpeduncular heterotopia to designate the interpeduncular structure based on its isointensity to normal parenchyma, a location not normally occupied by parenchyma, and lack of imaging evidence for a neoplastic or vascular process. We did not use the term hamartoma—though hamartoma and heterotopia may both be isointense to cortex—as a proportion of hamartomas is slightly T2-hyperintense relative to cortex. We nevertheless realize that without histologic confirmation the terminology of this presumedly malformative lesion is somewhat arbitrary.
Imaging findings reported in JSRD encompass the typical molar tooth sign and a range of additional CNS changes. These changes include cystic, Dandy-Walker–like enlargement of fourth ventricle and posterior fossa4 (as in patient 3), the rare occipital encephalo- or meningocele,5 abnormalities of the corpus callosum,1,6,7 myelination delay (as in patient 1), and unspecific white matter hyperintensities7,8 as well as migrational abnormalities.3,9,10 Absence of the decussation of the superior cerebellar peduncles and of the pyramidal tracts as well as lateralization of deep cerebellar nuclei can be demonstrated by using diffusion tensor imaging3 and is consistent with histopathology reports.11,12
Imaging findings in our patients were consistent with those reported previously in JSRD with the exception of the interpeduncular heterotopia. This finding has not been described previously, either in JSRD or as a separate entity. Although sagittal and 3D sequences provided greater morphologic detail, the interpeduncular heterotopia was already conspicuous on standard axial images and awareness of its possible occurrence seems the most likely means of increasing its detection. Interestingly, an interpeduncular structure similar to that in our patients is depicted, but not commented on in a paper on brain stem and cerebellar findings in Joubert syndrome (Fig 1c in Alorainy et al13). Whether interpeduncular heterotopia identifies a subset of patients with a specific geno- or phenotype cannot be inferred from our 3 patients. As of yet, no correlation between imaging findings and phenotype or genotype has been identified in JSRD, and there is considerable intrafamilial variability.5,14 In this context, absence of interpeduncular heterotopia in the affected sibling of patient 2 was not surprising.
The main differential diagnosis of the interpeduncular structure is that of hypothalamic hamartoma extending into the interpeduncular fossa. Hypothalamic hamartoma may occur in a specific form of JSRD, namely, in oral-facial-digital syndrome VI. Moreover, hypothalamic hamartoma in combination with the molar tooth sign is specific for oral-facial-digital syndrome VI.15 In our patients the interpeduncular lesion was clearly not of hypothalamic origin, nor did our patients present with the typical clinical findings of oral-facial-digital syndrome VI, namely, polydactyly, lingual hamartoma, and intraoral frenula.
A reported case of hypothalamic hamartoma without direct connection with the hypothalamus16 differed from the interpeduncular lesions in our patients by the lack of a broad contact to the mesencephalon, T2-hyperintensity relative to cortex, and a macroscopically cystic area.
The differential diagnosis further includes exophytic diencephalic and mesencephalic neoplasms, oculomotor schwannoma or cavernoma, meningioma, metastasis, pituitary macroadenoma, aneurysm, lipoma, epidermoid, and arachnoid cysts. Most of these entities can be differentiated from the interpeduncular lesions in our patients by their origin and by their signal intensity, if not already by their expansive and/or infiltrative nature. A further potential differential diagnosis is leptomeningial glioneural heterotopia. However, these microscopic pial buds or nests of disorganized glioneuronal tissue within the subarachnoid space17 are discovered on histopathology, and to our knowledge MR imaging has not been reported.
Pathogenetically, we believe that the interpeduncular heterotopia is part of a spectrum of malformative brain stem changes occurring in JSRD. Postmortem histopathology of 2 patients with Joubert syndrome has shown that there are extensive maldevelopmental changes in the brain stem, eg, hypoplasia of inferior olivary nuclei, disorganized tegmental gray matter, reduction of neurons in brain stem nuclei, and aberrant transverse pontine fibers.11,12 Together with the absent or partially absent decussation of the superior cerebellar peduncles and the pyramidal tracts,11,12 these changes indicate aberrant development and more specifically aberrant migration.
The mechanisms resulting in the abnormal brain development of JSRD are still incompletely understood, though identification of currently 10 causative genes has provided some insight: All genes implicated in JSRD encode for proteins involved in the formation or function of the primary cilium.18 Primary cilia are membrane-bound structures that project from the cell surface. They sense mechanical and chemical signals from the environment and modulate signaling pathways (eg, sonic hedgehog, WNT, platelet-derived growth factor α), which are important for tissue development and homeostasis. Sonic hedgehog signaling and cilium function are required for normal specification of the mid-hindbrain boundary and dorsal-ventral patterning. The malformations in JSRD are thought to result from defective dorsal-ventral patterning and cranio-caudal segment specification in the mid-hindbrain boundary as a consequence of the underlying defects of primary cilia.19 It is therefore tempting to speculate that the interpeduncular heterotopia in our patients is one manifestation of a spectrum of malformative brain stem changes occurring in JSRD and that it results from misdirected migration due to the underlying ciliopathy.
Conclusions
Interpeduncular tissue formation resembling heterotopia presents a previously undescribed, distinct imaging finding occurring in JSRD. Although the potential clinical and genetic implications of this finding remain to be elucidated, it seems likely that interpeduncular heterotopia is one manifestation of a spectrum of brain stem malformations in JSRD.
Footnotes
Disclosures: M.B., Research Support (including provision of equipment and materials): German Cancer Aid, Details: Research support for one fellow for 1 year; Speaker Bureau: Siemens, Codman, Micrus, Details: 1000€ each; Consultant: Codman, Details: 1000€
References
- Received October 8, 2010.
- Accepted after revision November 19, 2010.
- Copyright © American Society of Neuroradiology
In this issue









{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Ahi1 promotes Arl13b ciliary recruitment, regulates Arl13b stability and is required for normal cell migration
- Anterior Mesencephalic Cap Dysplasia: Novel Brain Stem Malformative Features Associated with Joubert Syndrome
- Joubert syndrome: neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause
- Novel asymptomatic CNS findings in patients with ACVR1/ALK2 mutations causing fibrodysplasia ossificans progressiva
- Joubert Syndrome and Related Disorders: Spectrum of Neuroimaging Findings in 75 Patients