
Abstract
BACKGROUND AND PURPOSE: The olfactory apparatus, consisting of the bulb and tract, is readily identifiable on MR imaging. Anomalous development of the olfactory apparatus may be the harbinger of anomalies of the secondary olfactory cortex and associated structures. We report a large single-site series of associated MR imaging findings in patients with olfactory anomalies.
MATERIALS AND METHODS: A retrospective search of radiologic reports (2010 through 2014) was performed by using the keyword “olfactory”; MR imaging studies were reviewed for olfactory anomalies and intracranial and skull base malformations. Medical records were reviewed for clinical symptoms, neuroendocrine dysfunction, syndromic associations, and genetics.
RESULTS: We identified 41 patients with olfactory anomalies (range, 0.03–18 years of age; M/F ratio, 19:22); olfactory anomalies were bilateral in 31 of 41 patients (76%) and absent olfactory bulbs and olfactory tracts were found in 56 of 82 (68%). Developmental delay was found in 24 (59%), and seizures, in 14 (34%). Pituitary dysfunction was present in 14 (34%), 8 had panhypopituitarism, and 2 had isolated hypogonadotropic hypogonadism. CNS anomalies, seen in 95% of patients, included hippocampal dysplasia in 26, cortical malformations in 15, malformed corpus callosum in 10, and optic pathway hypoplasia in 12. Infratentorial anomalies were seen in 15 (37%) patients and included an abnormal brain stem in 9 and an abnormal cerebellum in 3. Four patients had an abnormal membranous labyrinth. Genetic testing was performed in 23 (56%) and findings were abnormal in 11 (48%).
CONCLUSIONS: Olfactory anomalies should prompt careful screening of the brain, skull base, and the pituitary gland for additional anomalies. Genetic testing should be considered.
ABBREVIATIONS:
- CHARGE
- Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities (hypogonadism), Ear anomalies and/or deafness
- OB
- olfactory bulb
- OT
- olfactory tract
Olfaction involves recognition of myriad chemicals in various concentrations enabling discrimination among scents. In humans, the olfactory system provides less survival advantage than the other senses and the olfactory apparatus is proportionately much smaller than that in other mammals but is readily identifiable on routine MR imaging.1
The olfactory system begins to form early in gestation and has multiple origins, including the olfactory placode, cranial neural crest, and the olfactory bulb (OB), which is an extension of the telencephalon. The olfactory placode develops into the olfactory epithelium and nerves, which migrate toward the developing OB. Olfactory ensheathing cells are likely neural crest derivatives, which serve as a scaffolding for initial migration.2 Adjacent to the olfactory placode is the developing adenohypophyseal placode. The OB projection neurons or olfactory tracts (OTs) innervate multiple cortical regions and are collectively referred to as the olfactory cortex. The olfactory cortex comprises the anterior olfactory nucleus, olfactory tubercle, piriform cortex, entorhinal cortex, and septal, habenular, and brain stem nuclei as well as the amygdale, hippocampus, and parahippocampal gyrus.2,3
Thus, anomalies of the primary olfactory system might be associated with diverse and widespread anomalies of the remainder of the brain. Syndromes associated with malformed primary olfactory pathways include Kallmann syndrome; CHARGE syndrome (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities [hypogonadism], Ear anomalies and/or deafness); septo-optic dysplasia; and craniotelencephalic dysplasia, malformations for which the molecular genetics are variably defined.3,4 We reviewed a sizable population of patients with abnormalities of the primary olfactory pathways to further categorize the clinical presentation, syndromic associations, and genetic results and expand the range of associated brain malformations and provide correlative diffusion tensor imaging and results of genetic testing.
Materials and Methods
The study was approved by the Institutional Internal Review Board (University of Texas Southwestern Medical Center) and is Health Insurance Portability and Accountability Act–compliant. A retrospective search of radiologic reports was performed at a tertiary pediatric hospital by using the keyword “olfactory” with inclusive dates of January 2010 to December 2014. Consensus was reached about findings on MR imaging after review by 2 pediatric neuroradiologists with Certificates of Added Qualification in neuroradiology, each with ≥18 years' experience.
Inclusion criteria were diagnostic-quality coronal T2WI of ≤4-mm section thickness and absence or hypoplasia of the primary olfactory pathways. Patients with regional masses or parenchymal destruction were excluded. The medical record was reviewed for clinical presentation, clinical and laboratory evidence of neuroendocrine function, hearing loss, and additional cranial nerve dysfunction. Genetic results including chromosomal microarray, fluorescence in situ hybridization, karyotype, and any potential syndromic associations, were noted.
The OBs and OTs were qualitatively classified as normal, absent, or hypoplastic. Bulbs classified as hypoplastic lacked the typical anterior focal expansion at the level of the posterior crista galli. In cases of asymmetric OB or OT, the small side was considered abnormal. The olfactory sulci were designated as normal, hypoplastic, absent, or dysplastic. A pattern search of supratentorial content focused on callosal morphology, the septum pellucidum, hippocampus rotation, and malformations of cortical development. The posterior fossa was evaluated for the mid- and hind brain malformations and skull base anomalies. The optic nerves, chiasm, and tracts were evaluated for hypoplasia or absence. The presence and position of the neurohypophysis were documented, along with the presence of the infundibulum. When possible, the structures of the membranous labyrinth and lower cranial nerves were evaluated. The directionally encoded color maps from diffusion imaging were reviewed in a subset of patients for anomalies of major commissural, association, and projection fibers.
Results
Forty-four patients with olfactory anomalies were found by using the specified inclusion criteria. Three patients were excluded due to hydranencephaly, large craniopharyngioma, and a frontal encephalocele, resulting in a cohort of 41 patients. The age range was 0.03–18 years with a mean age of 4.5 years. The male/female ratio was 19:22.
Olfactory Findings
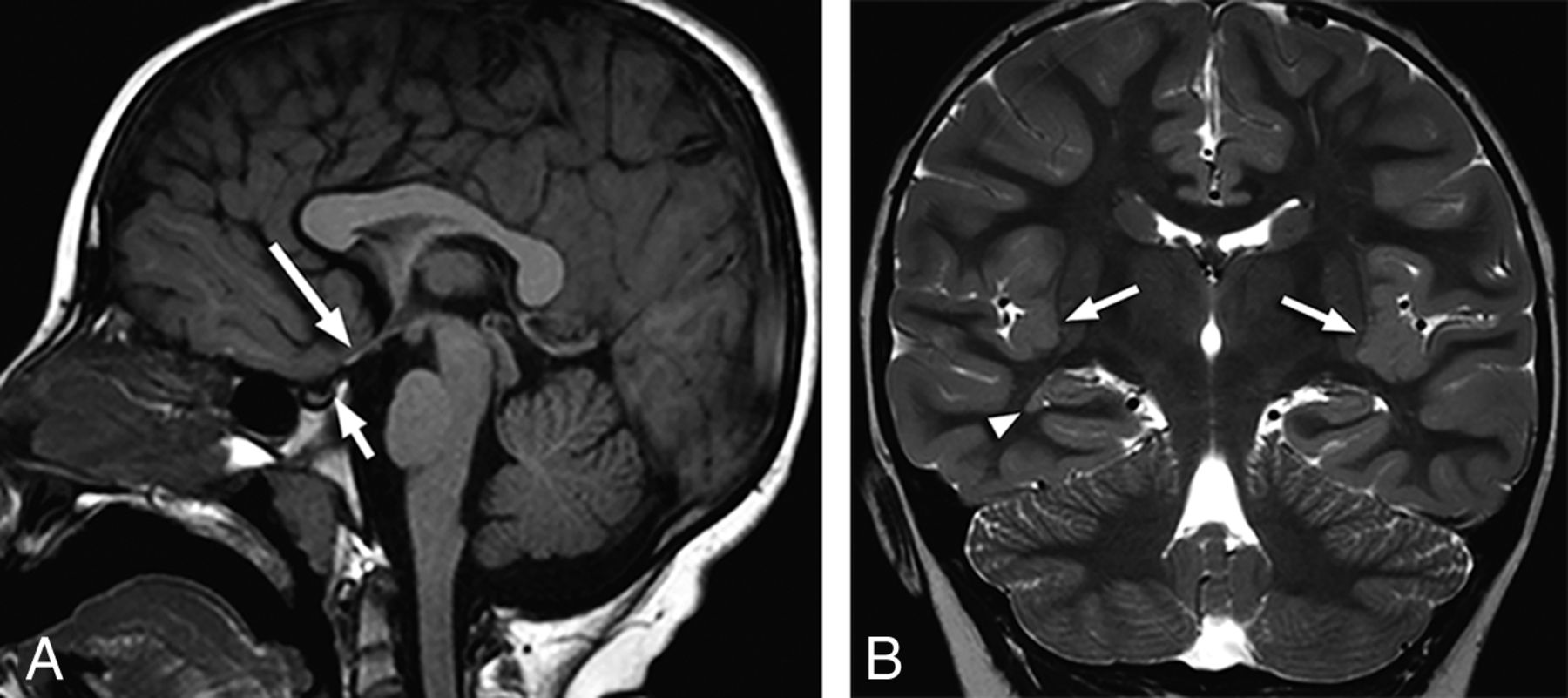
Of the 41 patients, abnormalities of the primary olfactory apparatus were bilateral in 31 (76%) and unilateral in 10 (24%). Fifty-six of 82 (68%) OBs and OTs were absent. Other olfactory findings included absent OB with OT hypoplasia in 5, hypoplastic OB with a normal OT in 5, absent OB with a normal OT in 2, hypoplastic OB and OT in 3, and a normal OB with a hypoplastic OT in 1. Sixty-one of 82 (74%) olfactory sulci were abnormal, and 38 were classified as hypoplastic. The sulcus was absent in 16 and dysplastic in 7. An abnormal sulcus was consistently associated with an abnormal OB and/or OT (Fig 1).

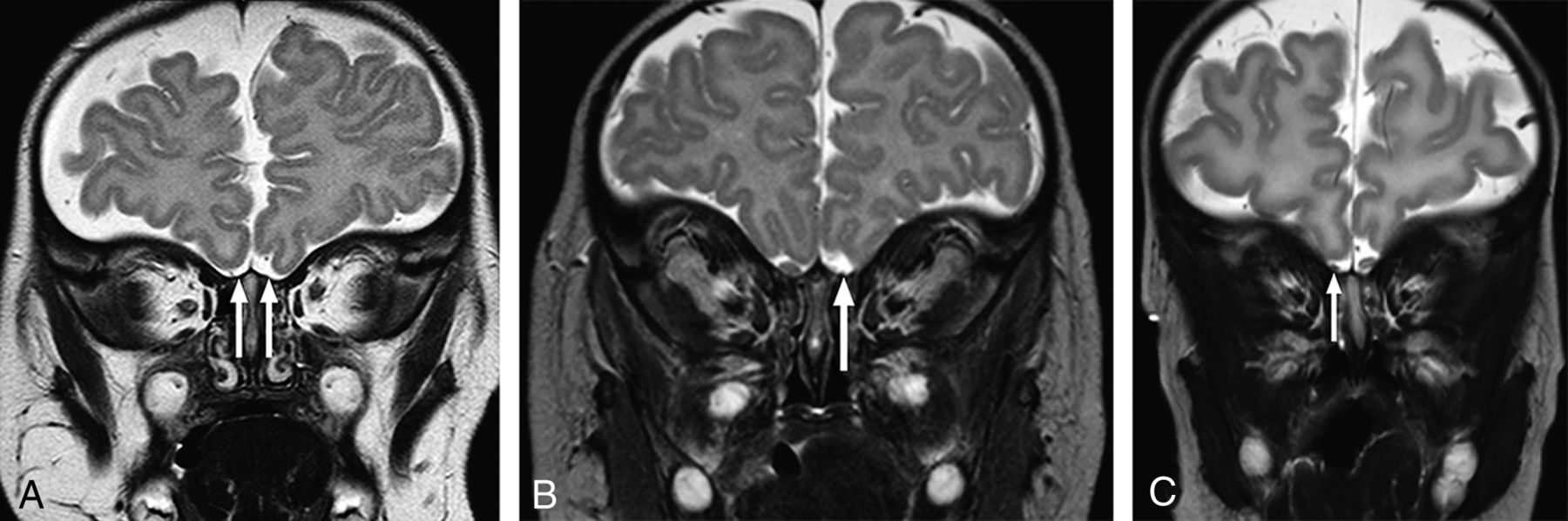
Coronal T2-weighted images at the expected region of the olfactory bulb demonstrating representative examples of olfactory anomalies found. A, There is bilateral absence of the OBs (arrows) with absence of the right olfactory sulcus. Small vessels are noted in the expected region of the olfactory OBs. No OTs are present (not shown). B, There is unilateral absence of the left OB (arrow) and OT (not shown). The olfactory sulcus is hypoplastic on the left. C, Note unilateral hypoplasia of the right OB (arrow) with a normal OT (not shown). The ipsilateral sulcus is hypoplastic.
Associated Imaging Abnormalities
Of the 41 patients, only 2 (5%) had no associated brain anomalies; 1 patient had an absent OB and OT bilaterally, and the other had bilateral OB hypoplasia. Anomalies seen in the other 39 (95%) patients included hippocampal dysplasia in 26/41 (63%), which was bilateral in 22 patients and unilateral in 4 (Fig 2). Fifteen (37%) patients had supratentorial cortical malformations. The most common cortical malformation was polymicrogyria involving the insular cortex and was found in 8 patients. Other malformations included pachygyria in 1, subependymal heterotopias in 4, transmantle heterotopias in 2, and thalamic fusion of varying degrees in 3 patients (Figs 3 and 4). The corpus callosum was dysgenetic in 10 (24%) patients, with complete agenesis in 4 (Fig 5).

A 2-year-old child with seizures and bilateral absent OBs and OTs. Coronal T2-weighted image (A) through the mid-temporal lobes demonstrates abnormal rotation of both hippocampi with laterally positioned fimbria (arrows). Midline sagittal T1-weighted image (B) shows hypoplasia of the ventral pons (arrow). Genetic information was not available.

A 1-month-old infant with dysmorphic features and hypothyroidism and absent ONs and OTs. Axial T2-weighted image shows thalamic fusion (arrow) and mild diffuse undersulcation. Note multiple chromosomal abnormalities on chromosomal microarray (Table, patient 9).

A 5-year-old child with seizures and absent OBs and OTs. Coronal T2-weighted image through the frontal lobes shows right transmantle cortical dysplasia (arrow).

A 3-year-old child with developmental delay and hypoplastic OBs and OTs. Midline sagittal T1-weighted image shows a thickened body of the corpus callosum (arrow) and poor definition of the pontomedullary junction (arrowhead). The occipital cervical junction is abnormal as well. A directionally encoded fractional anisotropy map confirms anomalous supracallosal fibers (not shown), which result in the enlargement of the callosal body. Trisomy 2p was found on chromosomal microarray.
Anatomic pituitary abnormalities were present in 11 of 41 (27%) patients, which included ectopic or absent neurohypophysis. The optic nerves, chiasm, and or tracts were absent, hypoplastic, or dysplastic in 12/41 (29%) patients, with associated septum pellucidum agenesis in 4. Ten of 12 (83%) patients with optic abnormalities had abnormalities of the pituitary gland. Additional intracranial abnormalities were found in 11 of 12 (92%) patients with optic abnormalities, most commonly peri-Sylvian polymicrogyria (Fig 6). Pituitary abnormalities were found in 2 patients without associated optic hypoplasia, both with ectopic neurohypophysis. Pituitary cysts were not considered abnormal.

A 6-year-old child with blindness, nystagmus, developmental delay, and absent OBs and OTs. Midline sagittal T1-weighted image (A) demonstrates hypoplasia of the optic chiasm (long arrow) and a small anterior pituitary gland with an absent neurohypophysis (short arrow). Coronal T2-weighted image (B) through the insular cortex shows subtle nodularity of the insular cortex bilaterally, consistent with polymicrogyria (arrows) and a small subependymal heterotopia adjacent to the right temporal horn (arrowhead). Imaging and clinical findings are consistent with optic hypoplasia syndrome with the septum pellucidum present. No abnormality was present on chromosomal microarray.
Five of 6 patients with sensorineural hearing loss had high-resolution MR imaging, which showed typical imaging findings of CHARGE syndrome, with absent semicircular canals and small vestibules in 3 (Fig 7). The cochlear nerve was deficient or absent in 4 ears, and the vestibular nerves were absent in 4 ears. One of these patients was found to have unilateral absence of the seventh cranial nerve as well. The 2 other patients had dilated vestibules with incorporation of the lateral semicircular canals, with 1 having enlarged cisternal segments of the fifth cranial nerve. Absent or hypoplastic third and fifth cranial nerves, both demonstrating a hypoplastic pons, were found in 2 children without sensorineural hearing loss.
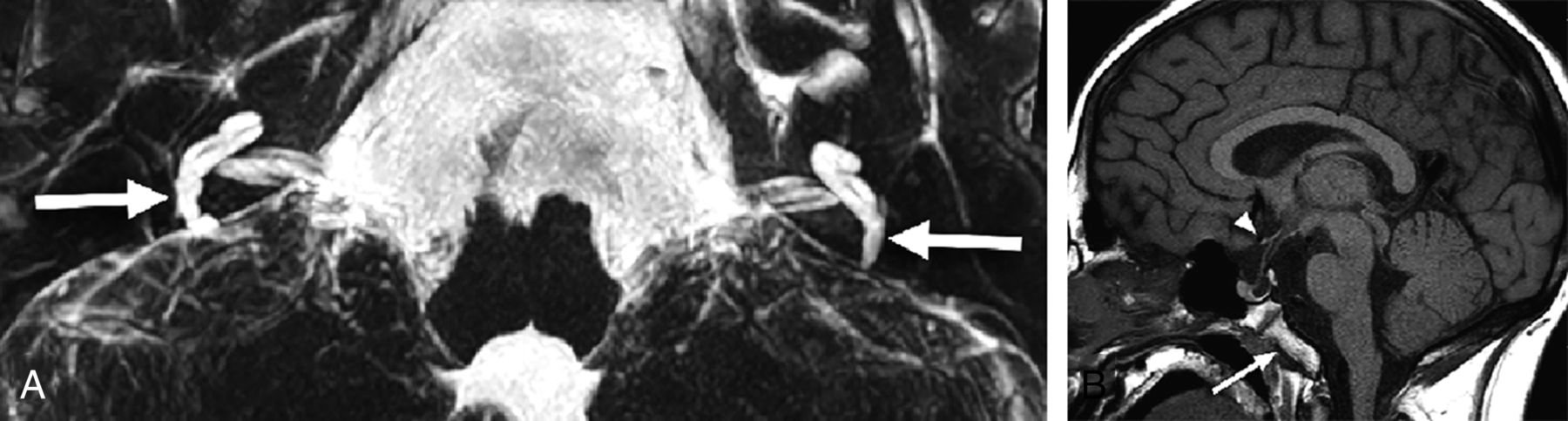
A 17-year-old adolescent with sensorineural hearing loss, panhypopituitarism, and absent OBs and OTs. Axial 3D T2-weighted MIP image (A) shows diminutive vestibules with vestigial posterior semicircular canals (arrows). No other semicircular canals are present. The cochlea is dysplastic bilaterally. Sagittal T1-weighted image (B) demonstrates an abnormal brain stem with an accentuated dorsal concavity and poor delineation of the ventral pontomedullary junction and a dysmorphic clivus (arrow). The optic chiasm is hypoplastic (arrowhead), and an anterior pituitary cyst is present. Imaging and clinical findings met criteria for CHARGE syndrome. Genetics information was not available.
Anomalies of the posterior fossa were found in 15 (37%) patients and were not usually isolated. Mid- and hind brain abnormalities included ventral pontine hypoplasia in 8 patients and an abnormal pontomedullary junction in 2 (Fig 2). Cerebellar vermian hypoplasia was seen in 3, and hemispheric dysplasia, in 1 (Fig 8). Four patients had a hypoplastic or dysplastic clivus with associated basilar invagination in 1 (Fig 7).
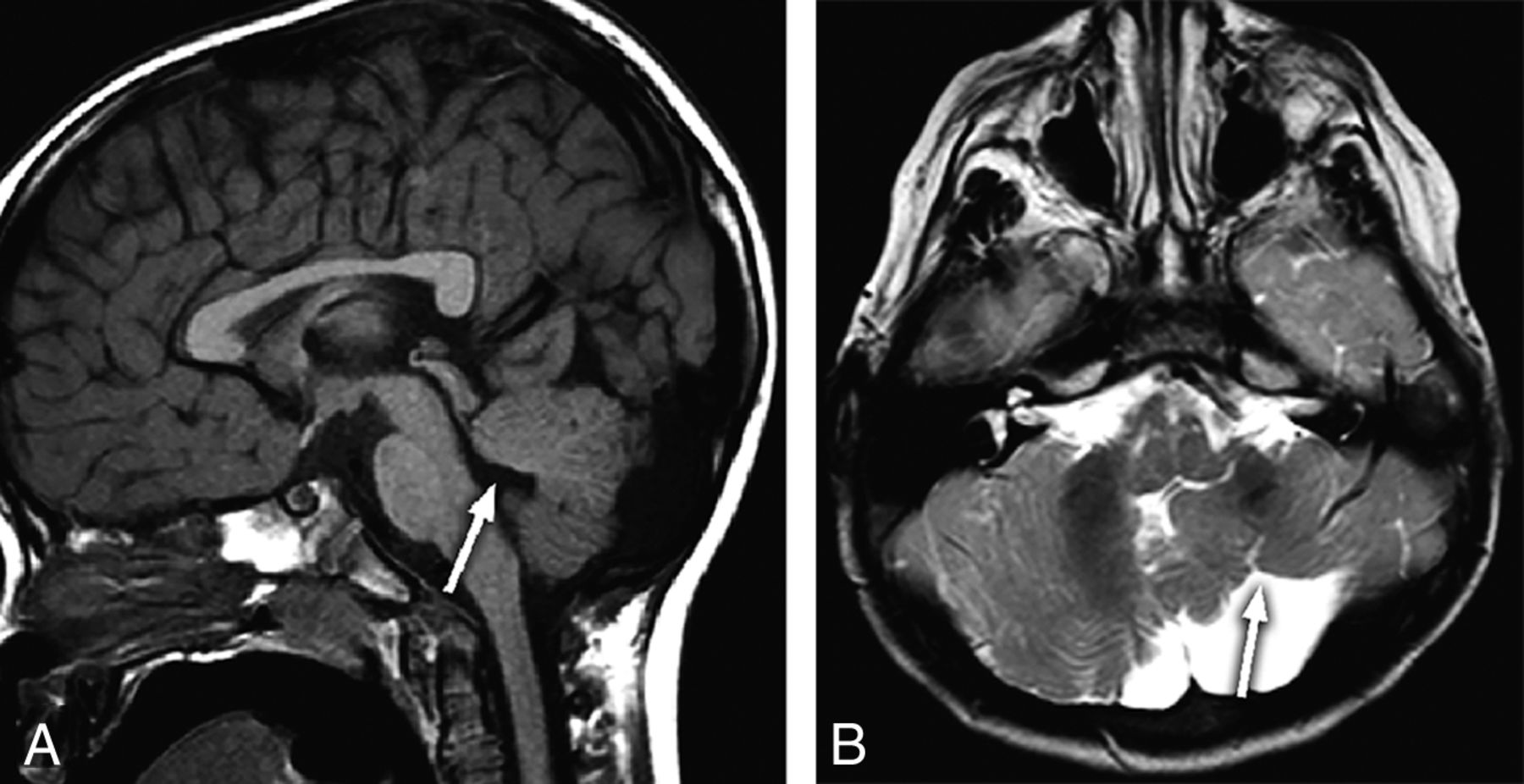
An 18-month-old child with developmental delay and an absent left OB and OT. Midline sagittal T1-weighted image (A) shows hypoplasia of the vermis with disorganization of the superior cerebellar lobules and deformity of the fourth ventricular roof (arrow). There is an incidental anterior pituitary cyst. Axial T2-weighted image (B) demonstrates a hypoplastic left cerebellar hemisphere with gyral disorganization (arrow).
Diffusion tensor imaging was available in 15 patients, and findings were abnormal in 8; 2 had complete callosal agenesis. Anomalous supracallosal fibers were seen in 1 patient with a thickened body of the corpus callosum (Fig 2). Anomalies of the brain stem on DTI included absence of the dorsal transverse pontine fibers in 3 patients and complete absence of transverse fibers in 1 patient (Fig 9). In this group, 1 patient had a normal pons by routine MR imaging but DTI showed a small corticospinal tract ipsilateral to left-sided basal ganglia hypoplasia and extensive bilateral frontal polymicrogyria.
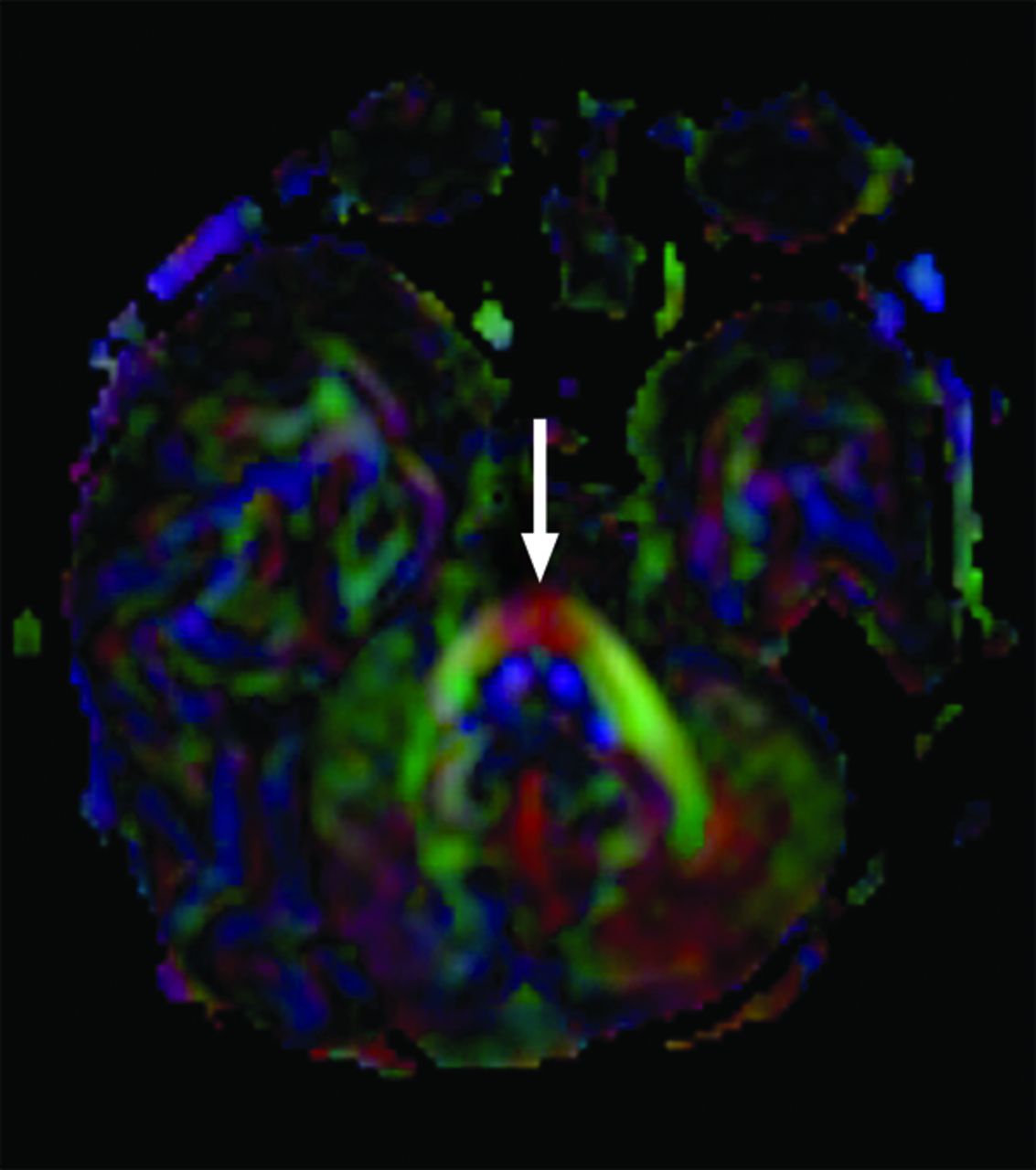
A 2-month-old infant with esotropia, seventh nerve palsy, and absent OBs and OTs. Axial directionally encoded fractional anisotropy map shows a single transverse pontine fiber bundle anterior to the corticospinal tracts (arrow). Anatomic images show only mild ventral pontine hypoplasia (not shown). Genetic information was not available.
Clinical Findings
The medical record was available for review in all patients. Twenty-four of 41 (59%) children were found to have developmental delay. Seizures were present in 14 of 41 (34%) patients, with coexisting developmental delay in 9. Neuroendocrine dysfunction was present in 14 of 41 (34%) patients, with 8 having panhypopituitarism; 2, isolated absent gonadotropin-releasing hormones; and 1, isolated growth hormone deficiency. Diabetes insipidus was present in 6 patients and was usually associated with anterior pituitary dysfunction. Pituitary hormone levels were evaluated in 18 of 41 patients. Pituitary function was considered normal in the absence of clinical indicators to suggest hormone deficiency or normal laboratory values. Eight patients had clinical findings indicating optic pathway pathology, including optic nerve hypoplasia and/or nystagmus, and sensorineural hearing loss was present in 6. Children commonly had multiple presenting symptoms (n = 25, 61%). Isolated anosmia or hyposmia was noted in only 3 patients.
Six children had multiple congenital anomalies reported, most commonly congenital heart disease, and 5 had dysmorphic facies. Cleft lip/palate or a bifid crista galli was noted in 4 patients. Syndromic associations were found in 9 (22%) and included CHARGE in 3, Kallmann in 2, as well as DiGeorge, Johanson-Blizzard, Jacobsen, and PHACE (posterior fossa malformations–hemangiomas–arterial anomalies–cardiac defects–eye abnormalities–sternal cleft and supraumbilical raphe) syndrome. A history of fetal alcohol syndrome or maternal drug exposure was present in 2 patients.
Genetic evaluation was performed in 23 (56%) patients, including chromosomal microarray in 19, fluorescence in situ hybridization in 3, and karyotype in 3, with some patients undergoing multiple genetic tests. Abnormal and diverse results were found in 11 (48%) patients (Table).
Abnormal genetic results
Discussion
A search of a large radiology report archive in a tertiary children's hospital netted 41 patients with confirmed abnormalities of the primary olfactory apparatus during a 5-year period, indicating that this malformation is uncommon. However, because detection of malformed OBs, OTs, and sulci requires coronal imaging, preferably T2, which may not be routinely performed, this spectrum of malformations is presumably more common than we appreciated. In our cohort, review of the medical records showed that most patients had no clinical symptoms referable to the olfactory pathway and usually presented with developmental delay (59%) and or seizures (34%). Neuroendocrine dysfunction (34%) was often associated as well. The myriad intracranial malformations associated with anomalies of the OB and/or OT may be explained by the extensive known anatomic and functional connections.3 Pachygyria, polymicrogyria, subependymal heterotopias, transmantle cortical dysplasia, and thalamic fusion were all found in this group of patients. There are multiple case reports and small series of olfactory anomalies either isolated or with associated abnormalities5⇓⇓–8; however, this is largest pediatric cohort reported.
The lateral projections or stria of the OTs extends to the parahippocampal cortex, with the region likely responsible for the characterization and memorization of odors. This feature may explain the high incidence of abnormal hippocampi seen in patients with olfactory anomalies. The presence of abnormalities of the brain stem may be predicted by the anatomic connections of medial projections of the OTs that extend to multiple locations within the brain stem, including the reticular formation, salivary nuclei, dorsal nucleus of cranial nerve X, and the olfacto-hypothalamo-tegmental bundle. Also, the development of the olfactory system occurs very early in gestation, suggesting the potential association with anomalies of phylogenetically older structures such as the posterior fossa.3 Potentially linking the formation of the cerebellar vermis and olfactory apparatus is the transcription factor zinc finger protein 423, which has been shown to be a requirement for patterning the development of precursors in the cerebellar vermis and olfactory apparatus in rats.9
In this cohort, agenesis of the corpus callosum was commonly found associated with olfactory anomalies. Abnormalities of the corpus callosum have been linked to OB absence, and this may be related to an absent induction phenomenon, with the olfactory bulbs representing a trigger for callosal development. In fetal mice, agenesis of the corpus callosum has been shown to result from destruction of the olfactory bulb.10 PAX6 mutations may result in agenesis of the OBs and corpus callosum as well as polymicrogyria.8 Also, TUBA1A mutations have been reported associated with absent OBs and agenesis of the corpus callosum and extensive brain anomalies, including dysplastic basal ganglia/thalami, cerebellar hypoplasia, and lissencephaly.11,12
One of the first reported clinical finding associated with congenital olfactory anomalies was isolated hypogonadotropic hypogonadism with absent or incomplete puberty, usually in males, in the setting of Kallmann syndrome. The gonadotropin receptors migrate along the olfactory tracts in the early fetus, explaining the association between low follicular stimulating and luteinizing hormones and absent olfactory bulbs and tracts.5,13 In our cohort, a minority of patients had isolated hypogonadotropic hypogonadism consistent with Kallmann syndrome. Panhypopituitarism was by far the more common pituitary presentation. Multiple genetic loci for Kallmann syndrome have been found, including the X-linked gene KAL1 and autosomal transmission on FGFR1 (chromosome 8), FGF8 (chromosome 10), PROKR2 (chromosome 3), and PROK2 (chromosome 20) genes.14,15 The PROKR2 and PROK2 mutations have also been reported to be the cause of isolated congenital anosmia.16 The 2 patients with suspected Kallmann syndrome did not have a chromosomal microarray.
Septo-optic dysplasia has been reported associated with olfactory bulb and tract hypoplasia.4 Ten of our patients with olfactory anomalies had imaging findings consistent with septo-optic dysplasia, with the entity defined in patients fulfilling 2 of 3 criteria (optic hypoplasia, pituitary dysfunction, and absent septum pellucidum). Two patients had imaging evidence of optic hypoplasia without additional criteria present. Most interesting, 6 of 10 patients with septo-optic dysplasia had a normal septum pellucidum. In the recent literature, the term “optic nerve hypoplasia syndrome” has been proposed as a more appropriate name because the development of the septum pellucidum is likely an independent process. The more consistent association with optic hypoplasia is cortical dysgenesis and hypopituitarism.17,18 Pituitary dysfunction was routinely identified (83%) in this subset of patients. Most interesting, a genetic link may be present between Kallmann syndrome and optic hypoplasia syndrome with loss-of-function mutations in PROKR2 found in both.19 The association of anterior pituitary dysfunction may relate to the proximity of the developing adenohypophyseal and olfactory placodes.2 Brain MR imaging protocols may benefit from the routine addition of coronal T2-weighted imaging, which would be optimal for evaluation of the olfactory apparatus and optic nerves.
Six patients had sensorineural hearing loss associated with olfactory anomalies. CHARGE syndrome was suspected 3 patients with typical inner ear malformations, including a small vestibule and absent semicircular canals. The olfactory nerves should be carefully evaluated in children with sensorineural hearing loss, especially with imaging findings consistent with CHARGE syndrome.6 The presence of anosmia in patients with CHARGE syndrome can be predictive of associated pituitary dysfunction; however, this association may be difficult to clinically assess in young patients and the presence of olfactory anomalies should prompt an evaluation of pituitary function.20,21 Panhypopituitarism was present in one of the patients with CHARGE. Basioccipital hypoplasia has also been reported to be common in CHARGE syndrome, with the CHD7 gene thought to have a role in neural crest development.22 Two of the patients with CHARGE syndrome had an abnormal clivus, and all 3 had dysmorphic brain stems. The olfactory nerves, regional facial structures, and skull base are derived from the cranial neural crest, and the association may explain the incidence of facial and skull base abnormalities present in this patient population.2 Absent semicircular canals and olfactory anomalies have also been reported in Waardenburg syndrome with SOX10 mutations, but this syndrome is typically associated with large vestibules. SOX10 is a regulator of neural crest development, which offers an additional link between normal development of neural crest structures and the olfactory apparatus.23,24
Additional chromosomal abnormalities have been reported associated with olfactory anomalies. In a recent report, a 6q27 microdeletion was present in 2 patients with olfactory bulb aplasia.7 One patient in our series had this microdeletion and associated ventral pontine hypoplasia. Partial trisomy (13 and 18) has been reported with olfactory aplasia, and these mutations were found in 2 of our patients.25 A 10q25 duplication was present in 1 patient and is in close proximity to the 10q24.32 gene mutation reported in the FGF8 type of Kallmann syndrome. DiGeorge syndrome is associated with a 22q11.2 deletion, with the findings of the fluorescence in situ hybridization analysis positive in our patient. A diverse clinical spectrum has been demonstrated with the syndrome.26,27 There are multiple reports linking the DiGeorge genetic abnormality to absent olfactory bulbs.28,29
The patients with Johanson-Blizzard syndrome had a confirmed UBR1 gene mutation as well as a 13q12.12 gain. The gene encodes for a protein involved in many basic biologic functions including neurogenesis and has been associated with absent OBs.30 The patient with Jacobsen syndrome had an 11q23.3 deletion and additional genetic abnormalities. Patients typically have facial dysmorphism, thrombocytopenia, and multiple malformations that include the CNS. Pachygyria, subependymal heterotopias, ventriculomegaly, and agenesis of the corpus callosum have been reported.31,32 Our patient had vermian hypoplasia, thalamic fusion, and optic hypoplasia. More than 40% of the 856 olfactory receptor genes are located along chromosome 11, possibly explaining the association, but to our knowledge, OB absence has not been reported in this syndrome. TUBA1A mutations were not found in our cohort but have been reported associated with absent OBs.
Fetal alcohol exposure has been reported to lead to abnormal olfactory development in adult mice, and impaired odor discrimination has been seen in children with heavy prenatal alcohol exposure.33,34 Our 2 cases of fetal alcohol and drug exposure may represent an in utero toxic acquired etiology rather than a genetic origin.
Diffusion tensor imaging has been reported to be helpful in evaluating the brain stem to determine the location and integrity of different white matter tracts.35 Diffusion tensor imaging found additional or clarified associated intracranial abnormalities in many of our patients. Morphologically abnormal brain stems were demonstrated in 10 patients, with absence or fusion of the transverse pontine fiber tracts often found in these patients. Asymmetric association tracts were identified, with associated thickened corpus callosum and prominent hippocampal commissures in children with agenesis of the corpus callosum. Diffusion tractography has been shown to be helpful in distinguishing the white matter connections within the corpus callosum, such as the presence of homo- or heterotopic fiber tracts.36,37
This was a retrospective study with resultant limitations inherently present. We cannot infer the incidence of olfactory anomalies in the pediatric population. The patients did not undergo a uniform clinical evaluation, and genetic evaluation was performed in just over one-half of the patients. All patients were not evaluated by an endocrinologist and did not undergo laboratory evaluation of pituitary function. Imaging was not specifically tailored to visualization of the OBs and OTs; however in our experience, the structures are adequately visualized on standard 4-mm coronal T2-weighted images. Olfactory bulb hypoplasia designation was subjective but was agreed on by consensus.
Conclusions
Children with OB and OT anomalies rarely present with symptoms referable to diminished smell. While pituitary dysfunction was a typical presentation in patients with olfactory anomalies, panhypopituitarism, not isolated hypogonadotropic hypogonadism, was the most common presentation. A wide spectrum of neurologic presentations was found, with seizures and developmental delay most common. Two distinct known associations of optic nerve hypoplasia syndrome and sensorineural hearing loss were commonly encountered. However, novel syndromic and genetic associations were also found. Anomalies of the olfactory apparatus are indicators of the presence of pituitary dysfunction and additional and often multiple brain malformations, with coronal T2-weighted imaging required for diagnosis. The results of genetic testing in this group are commonly abnormal and are suggested in the evaluation.
Footnotes
Paper previously presented at: Annual Meeting of the American Society of Neuroradiology and the Foundation of the ASNR Symposium, April 25–30, 2015; Chicago, Illinois.
References
- Received November 19, 2015.
- Accepted after revision January 14, 2016.
- © 2016 by American Journal of Neuroradiology









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}