
Abstract
SUMMARY: Galactosemia is a rare genetic condition caused by mutation of enzymes involved in galactose and glucose metabolism. The varying clinical spectrum reflects the genetic complexity of this entity manifesting as acute neonatal toxicity syndrome, requiring prompt diagnosis and treatment, to more insidious clinical scenarios as observed in the subacute and chronic presentations. The current literature predominantly focuses on the long-standing sequelae of this disease. The purpose of this multicenter clinical report comprising 17 patients with galactosemia is to highlight the MR imaging patterns encompassing the whole spectrum of galactosemia, emphasizing the 3 main clinical subtypes: 1) acute neonatal presentation, with predominant white matter edema; 2) subacute clinical onset with a new finding called the “double cap sign”; and 3) a chronic phase of the disease with heterogeneous imaging findings. The knowledge of these different patterns together with MR spectroscopy and the clinical presentation may help in prioritizing galactosemia over other neonatal metabolic diseases and prevent possible complications.
Lactose is the primary source of carbohydrates in neonates. The metabolism and sustaining energy supplementation are pivotal and are channeled through the Leloir pathway. Lactose is catabolized to galactose and glucose and eventually into uridine diphosphate galactose–galactose and uridine diphosphate galactose–glucose. Reduction of enzymatic activity results in an inborn error of metabolism manifesting as galactosemia. Deficiency of galactose-1-phosphate uridyltransferase is associated with the classic galactosemia phenotype or type I galactosemia (Online Mendelian Inheritance in Man, No. 230400; omim.org), an inherited autosomal recessive disorder caused by mutation of the galactose-1-phosphate uridyltransferase–encoding GALT gene on chromosome 9p13.3, with an estimated incidence of 1/30,000.1 Genetic mutations in the Leloir pathway involving galactokinase and uridine diphosphate galactose 4-epimerase cause type II and III galactosemia, respectively. The latter variants are rarer than classic galactosemia and manifest with insidious onset and a mild clinical course.2
The variable clinical outcome in type I galactosemia and disease severity is attributable to the polymorphic nature and molecular heterogeneity of galactose-1-phosphate uridyltransferase.3 Estimating erythrocyte activity of galactose-1-phosphate uridyltransferase remains the criterion standard technique in diagnosing classic galactosemia (type I), whereas galactokinase (type II) or the uridine diphosphate galactose 4-epimerase mutation (type III) enzymatic activity are measured in white blood cells.4,5
Galactosemia is an extremely complex disorder whose clinical spectrum depends on the interaction between genetic (ie, the residual activity of the affected enzyme) and environmental factors. Therefore, symptoms may vary from an acute and lethal presentation, frequently observed in the neonatal period, to more subacute or chronic forms whose pathogenesis has not been well-elucidated.3 Similarly, the diversity is also reflected in imaging features.
The chronic stage of the disease has been well-cited in the literature, whereas the data on acute neonatal presentation are limited to case reports.6,7 We intend to present a case series of 17 patients showcasing the divergent imaging patterns, ranging from the emergent neonatal toxicity syndrome, including its complications and sequelae, to the subacute presentations in young children and the more subtle alterations encountered in the chronic juvenile stage.
Case Series
Local institutional review board approval was obtained to analyze the clinical and laboratory records and imaging information of confirmed cases (Great Ormond Street Hospital, London, UK; Bambino Gesù Children’s Hospital, Rome, Italy; Hospital de Santa Casa de Misericórdia de São Paulo, São Paulo, Brazil). Clinical medical records from 17 patients (15 males and 2 girls; age range, 13 days to 14 years) from 3 pediatric hospitals were retrospectively reviewed.
All MR images were obtained on a 1.5 or a 3T scanner, which included sagittal and axial T1 spin-echo, axial and coronal T2-weighted, axial DWI, and axial or coronal FLAIR sequences. MR spectroscopy was also acquired in 7 patients with a single-voxel technique (TE = 35 ms).
Clinical and MR Imaging Findings at Presentation
Demographic, clinical, and imaging findings of the 17 patients are summarized in the Online Supplemental Data.
Acute Presentation.
Three patients (patients 3, 7, 10) initially presented at 13–25 days of life with macrocephaly and acute symptoms like vomiting, hypotonia, lethargy, jaundice, and variable severity of liver dysfunction. Patient 7 at day 12 of life developed septic shock with disseminated intravascular coagulation, and blood cultures were positive for Escherichia coli infection.
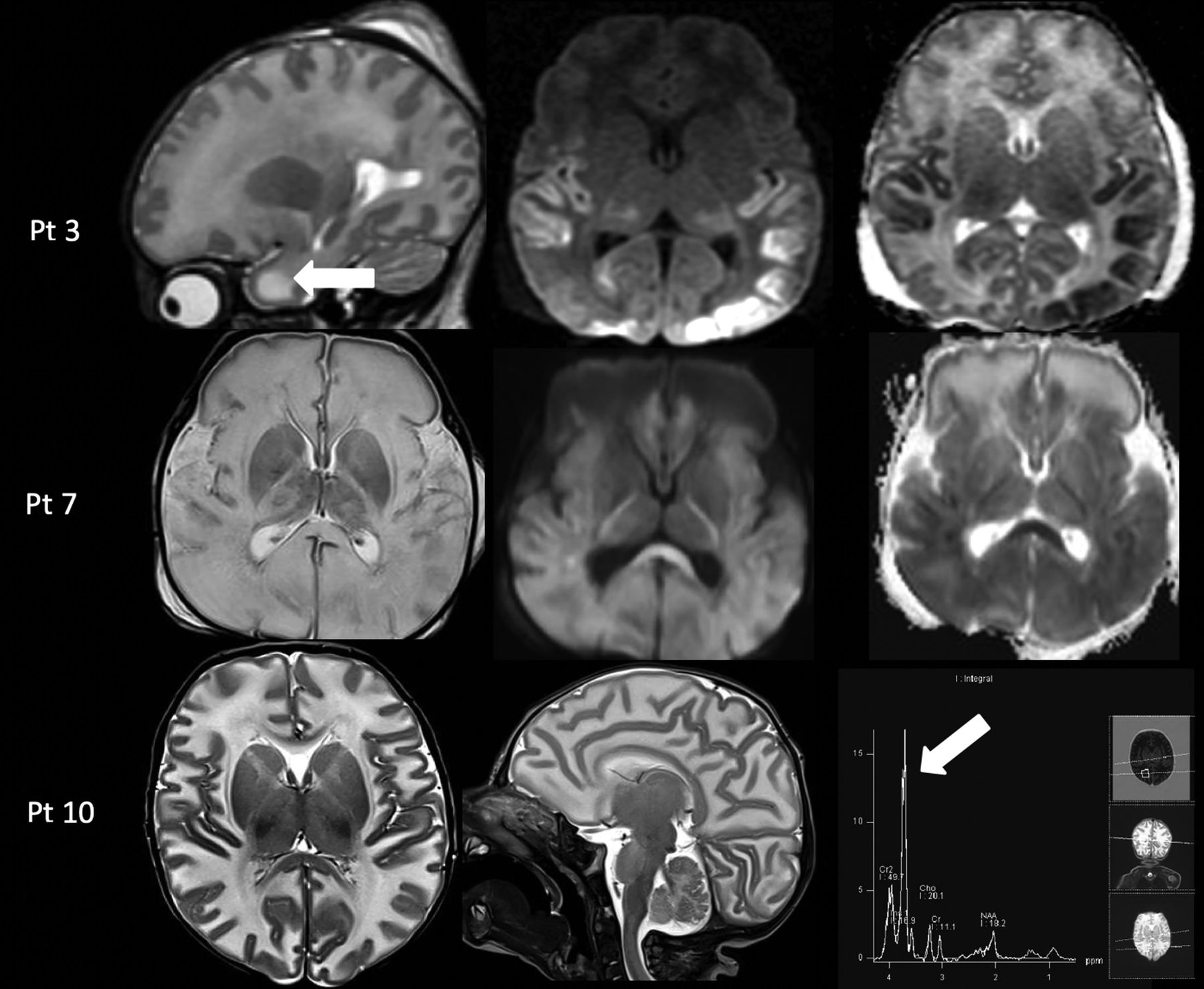
MR imaging demonstrated diffuse brain edema. Diffusion-restriction abnormality involving the temporo-occipital cortex (patient 3) and diffuse involvement of the deep white matter and corpus callosum (patient 7) were noted on the DWI sequence. Single-voxel MR spectroscopy was performed (patients 3 and 10) in the ROI and demonstrated the presence of an abnormal doublet peak at 3.67–3.74 ppm, representing galactitol (Fig 1).7

Acute toxicity syndrome in 3 cases of galactosemia. Upper row: MR imaging in a 13-day-old male infant (patient 3) with acute liver failure demonstrates diffuse white matter edema with the cystic appearance of the anterior temporal poles on a sagittal T2-weighted image (left column, arrow) and cytotoxic edema in the temporo-occipital cortex and thalami on the axial DWI and ADC maps (middle and right columns). Middle row: MR imaging in a 17-day-old male infant (patient 7) with profound hypotonia and sepsis showing diffuse white matter edema on T2-weighted image (left column) and diffuse cytotoxic edema on DWI and ADC maps (middle and right column). Lower row: MR imaging in a 25-day-old male infant (patient 10) with lethargy and vomiting shows diffuse white matter edema on axial and sagittal T2-weighted images (left and middle column) and an abnormal galactitol peak at 3.7 ppm on MR spectroscopy with a short TE of 35 ms (right column, arrow). Pt indicates patient.
Subacute Presentation.
Five patients (patients 1, 2, 15, 16, 17) presented between 7 and 18 months of age with a subacute clinical onset characterized by psychomotor retardation, developmental delay, cataracts, macrocephaly, hepatosplenomegaly, renal tubulopathy (patients 1 and 2), and seizures (patient 17).
In these patients, MR imaging showed diffuse white matter edema associated with thinning of the corpus callosum (according to the biometric values proposed by Garel et al8). A bean-shaped dark line was clearly observed on T2-weighted images with a central area of hyperintensity around the frontal horns of the lateral ventricles demonstrating a “double cap sign” appearance. This was better appreciated on FLAIR sequences in which the central area showed a hypointense signal. Additionally, cystlike lesions in the bilateral anterior temporal poles were observed. Single-voxel MR spectroscopy in the abnormal white matter was performed in 4/5 patients and revealed an abnormal doublet peak at 3.67–3.74 ppm, confirming the accumulation of galactitol (Fig 2). The thalami and anterior portions of the striatum were also symmetrically involved in patients 1 and 2 (Fig 3).

Subacute presentation. MR imaging of patient 15 at 8 months of age (A–E) demonstrates the presence of diffuse white matter edema on an axial T2-weighted image (A) associated with the double cap sign, best appreciated on T2 and axial FLAIR images (A and B, arrows). Sagittal T1-weighted image shows the presence of cystlike lesions of the temporal poles (C, arrow) and thinning of the corpus callosum (D, arrow). Single-voxel MR spectroscopy with a short TE of 35 ms demonstrates the presence of a doublet peak at 3.6–3.74 ppm, corresponding to galactitol (E, arrow). Follow-up MR imaging performed at 2 years of age (F–J) shows complete resolution of white matter edema on axial T2-weighted sequences (F); the double cap sign and temporal lobe cysts, respectively, on axial (G) and sagittal FLAIR (H), with the presence of patchy alterations in the periventricular white matter, especially in the frontal lobes (G and H, arrows); and thinning of the corpus callosum on sagittal T1-weighted image (I, arrow). MR spectroscopy demonstrates the disappearance of the previously noted galactitol peak (J).
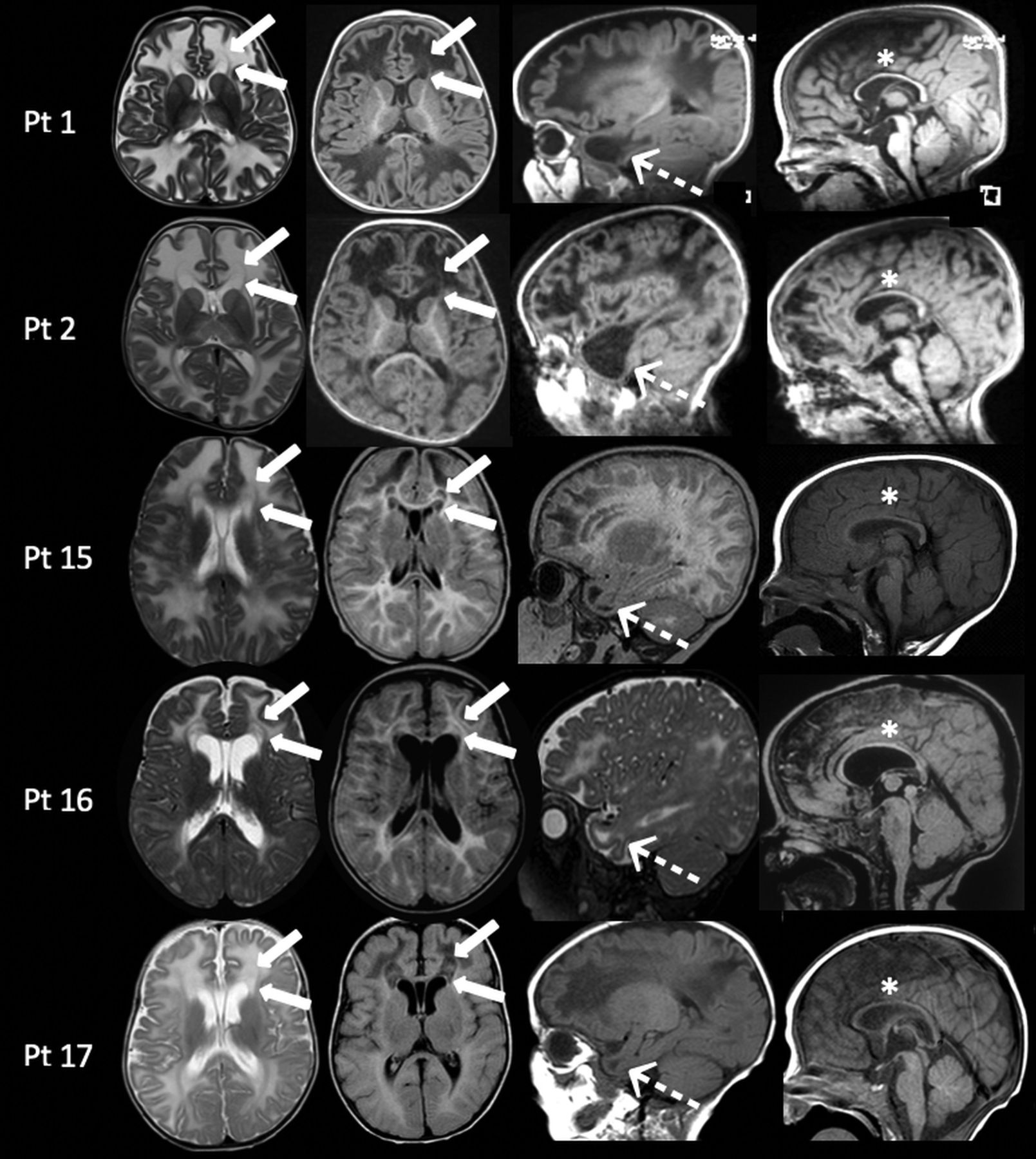
MR imaging findings in patients with galactosemia with subacute clinical presentations between 7 and 18 months of age. In all patients, axial images (left columns) show diffuse white matter edema associated with the presence of the double cap sign adjacent to the frontal horns of the lateral ventricles (arrows). On sagittal images (right columns), MR imaging demonstrates, in all patients, the presence of cystlike lesions of the temporal poles (dotted arrows) and thinning of the corpus callosum (asterisks). Pt indicates patient.
Chronic Presentation.
In the remaining 9 patients, MR imaging was performed later in life (between 2.11 and 14 years) during a galactose-free diet, and the clinical condition was characterized by varying degrees of intellectual disability associated with autism spectrum disorder (patient 6), dystonia (patient 5), and acute symptoms, including vomiting and seizures (patient 4). MR imaging demonstrated variable findings including the following: 1) in patients 4 and 13, the presence of multiple patchy areas of T2 hyperintensity in the supratentorial white matter; 2) delayed myelination with variable thinning of corpus callosum (patients 8, 9, 11, and 14); and 3) the absence of relevant imaging findings (patients 6 and 12). Patient 5, with severe dystonia, demonstrated symmetric T2 hyperintensity in the globus pallidus without significant alterations in the rest of the brain.
Clinical/Imaging Follow-Up
Clinical follow-up was available in 6 patients (patients 3, 4, 5, 7, 8, 15) at different ages (Online Supplemental Data). All of them demonstrated cognitive impairment. MR imaging was available at clinical follow-up in 5 patients with variable findings.
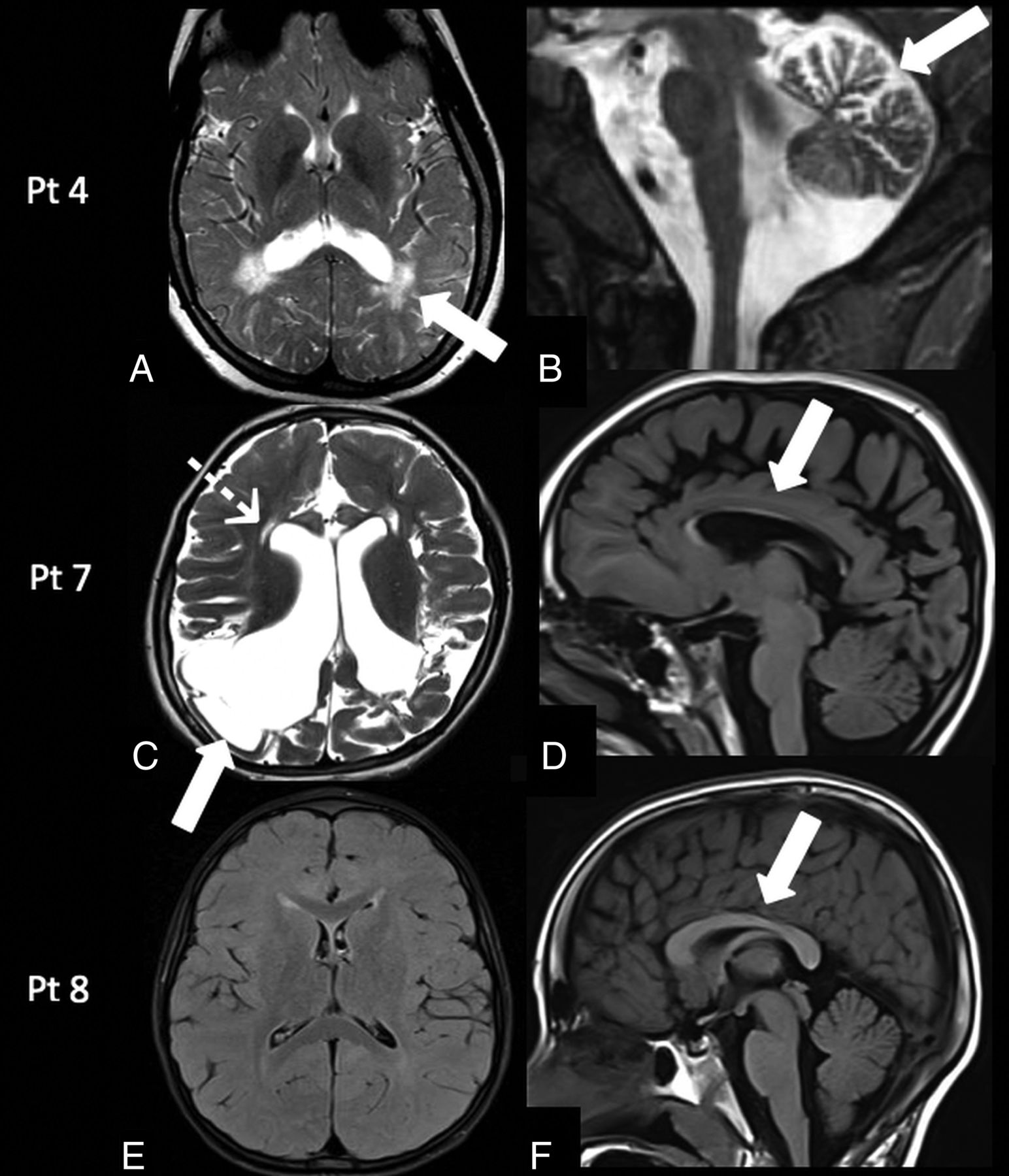
Patient 3 was reassessed at 2 years of age and had a language delay. Patient 4 at 13 years of age had mild cognitive impairment (learning difficulties), left-side weakness, ovarian failure, osteopenia, and epilepsy. MR imaging demonstrated persistence of a peritrigonal and subcortical white matter signal abnormality associated with mild cerebellar atrophy (Fig 4A, -B).
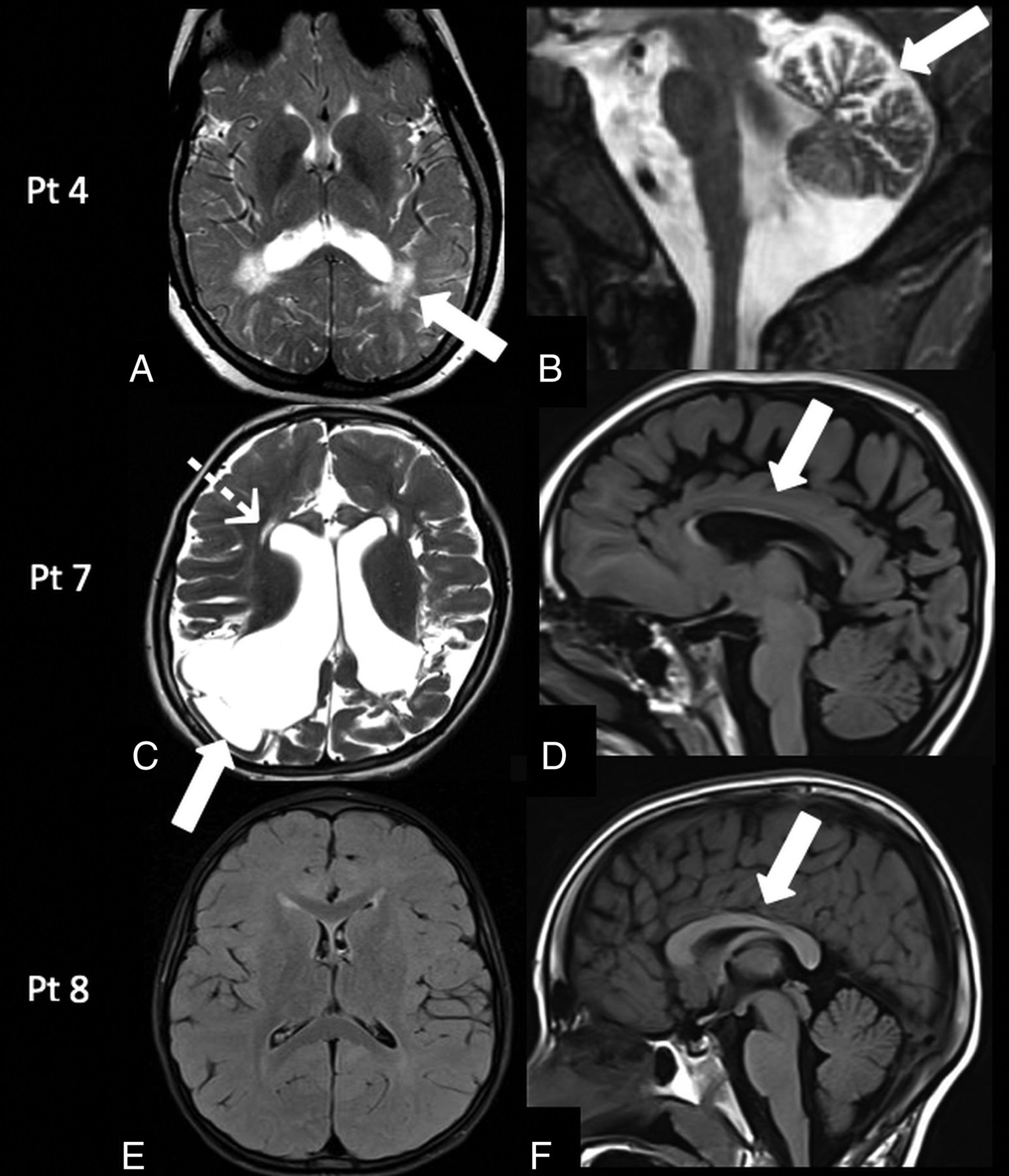
Follow-up MR imaging findings in galactosemia. Patient 4: MR imaging at 13 years of age shows the persistence of the peritrigonal white matter abnormalities on axial T2-weighted image (A, arrow) associated with the presence of mild cerebellar atrophy on sagittal T2-weighted image (B, arrow). Patient 7: MR imaging at 5 years of age demonstrates encephalomalacic changes in the bilateral cortical and subcortical parieto-occipital regions on axial T2-weighted image (C, arrow), with ventricular dilation, cystic changes in the periventricular white matter (C, dotted arrow), and corpus callosum thinning on a sagittal T1-weighted image (D, arrow). Patient 8: MR imaging at 7 years of age demonstrates persistently delayed myelination on an axial FLAIR image (E) and a normal corpus callosum (F, arrow). Pt indicates patient.
In patient 5, there was progressive worsening of dystonia, and male infant died at 11 years of age following severe respiratory failure due to bronchopneumonia.
Patient 7 at 5 years of age had microcephaly, motor stereotypies, self-injury behavior, spastic tetraparesis, and language paucity. MR imaging demonstrated the presence of cystic encephalomalacic changes in the bilateral parieto-occipital regions, significant reduction of white matter bulk resulting in thinning of the corpus callosum, and cystic changes in the periventricular frontal white matter (Fig 4C, -D).
Patient 8 at 7 years of age had a mild global developmental delay, and MR imaging confirmed persistent delayed myelination (Fig 4E, -F).
Patient 15 underwent follow-up MR imaging after dietary restrictions at 2 years of age, which demonstrated complete resolution of brain edema including the double cap sign and cystlike lesions in the temporal poles with patchy periventricular supratentorial white matter abnormalities. In addition, MR spectroscopy demonstrated posttreatment changes, with disappearance of the abnormal galactitol doublet (Fig 2).
DISCUSSION
Since the initial description of galactosemia in 1908, several pathogenic genetic mutations have been identified, and the awareness of the possible clinical presentation has significantly increased with time.1
Nelson et al,9 in 1992, reported the first systematic description of imaging findings highlighting the abnormal T2-hyperintense signal in the peripheral white matter, suggesting delayed myelination. Since then, only a few case reports have been published, mainly focusing on the acute presentation of the disorder.6,7
However, it is now evident that all forms of galactosemia present as a continuum disorder in which clinical symptoms depend on the interaction between genetic mutations and environmental factors.10 Due to lack of a precise pathophysiologic mechanism, galactosemia remains poorly understood, and several multifactorial hypotheses contribute to explaining the disorder, including accumulation of toxic metabolites, defects in galactosylation of glycolipids and glycoproteins, and myo-inositol depletion.11
In our series of 17 patients, 3 presented with neonatal toxicity syndrome, a medical emergency characterized by poor feeding, vomiting, bulging of the anterior fontanelle, lethargy, and hypotonia. Impaired activity of the galactose-1-phosphate uridyltransferase enzyme results in accumulation of toxic metabolites, namely galactitol, galactose-1-phosphate, galactonate, and galactose. Galactitol is an end product of the aldose reductase activity induced by elevated galactose levels.3 Galactitol is poorly diffusible and highly osmotic, and its intracellular accumulation contributes to cell swelling. This specifically occurs in vulnerable tissues like the lens, predisposing to cataract formation, and in the brain parenchyma, inducing cerebral edema with high mortality in untreated or undiagnosed cases.12
During the acute phase of the disease, MR imaging demonstrates diffuse brain edema with increased T2 signal in the white matter and areas of restricted diffusion involving the cortex and deep gray matter nuclei, consistent with cytotoxic edema. MR spectroscopy facilitates the identification of the galactitol doublet at 3.67 and 3.74 ppm, with reduction of myo-inositol-, choline-, and N-acetylaspartate-containing compounds (Fig 1).7 The increase in morbidity and mortality in the acute clinical setting can be further complicated by E coli sepsis, as seen in patient 7 of our study.13 Prompt initiation of a lactose-free diet along with effective measures to prevent sepsis and coagulopathy are crucial in the clinical outcome of neonatal toxicity syndrome.
In 5 patients of our series, the disease manifested later in life (between 7 and 18 months of age) with subtle presentation, mainly characterized by macrocephaly, hepatomegaly, and psychomotor developmental delay. MR imaging revealed diffuse brain edema, with a peculiar imaging finding, the double cap sign, observed in the frontal lobes (Figs 2 and 3). To our knowledge, these findings have not been described in the imaging literature. They are typically better appreciated on FLAIR images as a darker central zone attributable to accumulation of free water without diffusion restriction on DWI and suggests vasogenic edema within the fibers of the forceps minor. Reversibility of these changes on follow-up imaging was demonstrated, thereby reinforcing and supporting our assumption of vasogenic edema (Fig 2). In addition, cystlike lesions were also noted in the anterior temporal poles (Fig 3). Anterior temporal lobe cysts have been well-described as a hallmark of some congenital diseases and genetic leukoencephalopathies.14
We hypothesize that the observed findings may represent the effect of significant vasogenic edema within structures that are normally characterized by an increased free-water content (the deep white matter) and a more delayed myelination compared with other lobes.15 This hypothesis could also possibly explain why the FLAIR sequence, which is sensitive to both T1- and T2-relaxation times, may be particularly sensitive to demonstrate the double cap sign by inverting the CSF-like signal within edematous myelinating regions, with higher free-water content in the frontal lobes with partial sparing of myelinated fibers of the forceps minor and the periventricular bright line, normally described on FLAIR images before complete myelination.15 This finding may serve as a relevant imaging clue to prioritize galactosemia among the list of differential diagnoses. MR spectroscopy was not only pivotal in identifying the galactitol peak, but follow-up imaging also demonstrated resolution of the abnormal spectral pattern and thereby correlates well with initiation and maintenance of an appropriate dietary regimen in our patient (patient 15) (Fig 2).
Despite being on a galactose-free diet, patients may also experience delayed neurologic symptoms, which are believed to be the result of the endogenous production of galactose from glucose and the accumulation of secondary toxic products such as galactitol and galactose-1-phosphate.16 It is evident that early detection and avoiding complications of acute neonatal toxicity syndrome may prevent cataracts, but it does not alter the overall long-term neurologic morbidity.3 It is not well-understood whether these complications result from developmental abnormalities that are initiated in utero, long-term exposure to endogenously produced galactose, poor dietary control, or a combination of both and other undetermined factors.3,4 A prompt diagnosis with early treatment based on galactose dietary restriction is associated with considerable symptom improvement, though some degree of cognitive developmental delay persists, particularly in the language and executive function domains.17
In our case series, conventional MR imaging findings in a galactose-free dietary regimen demonstrated a lack of significant signal abnormalities (patients 6 and 12), patchy white matter signal alterations (patients 4, 13, and 15), and delayed myelination (patients 8, 9, 11, and 14). Ventricular enlargement and cerebellar atrophy were also noted, and our observations match those in the existing imaging literature.9 Although the exact mechanism of injury remains unclear, pathologic studies in the past have suggested the deficiency of galactocerebrosides as a possible cause of myelin breakdown.18 Impaired myelination has also been postulated from the results of a recent study investigating in vivo microstructural alteration in patients with galactosemia with advanced imaging techniques such as neurite orientation dispersion and density imaging.19
In our series, 1 patient (patient 5) presented with severe dystonia, and MR imaging findings at 7 years of age revealed bilateral globus pallidus abnormalities. These findings are atypical for classic galactosemia and may represent a sequela of neonatal kernicterus as reported by the clinical history of the patient. This case highlights the importance of possible comorbidities while evaluating patients affected by galactosemia.
The only efficacious treatment of classic galactosemia is a dietary galactose restriction, though the necessity to maintain a life-long restriction remains a matter of debate and is not sufficient to prevent long-term sequelae.2,20
In conclusion, galactosemia is a rare genetic disorder with a continuum spectrum of varying imaging and clinical profiles ranging from a life-threatening acute medical emergency to delayed cognitive effects. Conventional MR imaging features in the delayed phase are often nonspecific and overlap other pathologies. The double cap sign observed in our case series may help in prioritizing galactosemia over other neonatal metabolic diseases, with the added benefit that performing MR spectroscopy to detect a galactitol peak can prevent the complications of the acute neonatal toxicity presentation of galactosemia.
Footnotes
K. Mankad and L.L.F. do Amaral contributed equally to the article.
Disclosures: Kshitij Mankad—UNRELATED: Employment: I am paid a regular salary by Great Ormond Street Hospital; Expert Testimony: I offer private medicolegal reports; Payment for Lectures Including Service on Speakers Bureaus: speaker honorarium from Novartis and Siemens.
References
- Received July 21, 2020.
- Accepted after revision October 23, 2020.
- © 2021 by American Journal of Neuroradiology









{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.