
Abstract
Anaplastic large cell lymphomas (ALCL) are mature T-cell neoplasms, approximately half of which harbor rearrangements of the ALK gene that confer a good prognosis. Recent studies have demonstrated that a significant proportion of ALK-negative ALCLs demonstrate rearrangements of the IRF4/DUSP22 locus that also are typically associated with a favorable prognosis. ALCL with primary involvement of the central nervous system (CNS) is extremely rare. We report what may be the first case of ALK-negative ALCL with IRF4/DUSP22 rearrangement involving the brain in a 55-year-old man. Magnetic resonance imaging demonstrated signal abnormalities in the periventricular region, corpus callosum and cingulate gyrus. Biopsy revealed a diffuse parenchymal and angiocentric infiltrate of CD30-positive cells that showed IRF4/DUSP22 rearrangement by fluorescence in situ hybridization. We also review the clinical and pathologic features of primary CNS ALK-negative ALCLs in the literature and highlight the need for awareness of this entity to optimize appropriate management.
Similar content being viewed by others


Introduction
Anaplastic large cell lymphomas (ALCLs) are mature T-cell neoplasms expressing CD30, a lymphocyte activation marker [1,2,3]. They are heterogeneous genetically and clinically, and show a morphologic spectrum as well as overlap with other T-cell lymphomas, sometimes making diagnosis difficult [4,5,6]. According to the World Health Organization (WHO), there are four types of ALCLs, systemic ALK-positive ALCL, systemic ALK-negative ALCL, primary cutaneous ALCL, and the provisional entity breast-implant-associated ALCL [3, 4]. Approximately half of systemic ALCLs show translocations involving the anaplastic lymphoma kinase gene ALK on 2p23, which is associated with a good prognosis, partly due to ALK-positive ALCL being more common in children and young adults whereas ALK-negative ALCL peaks in middle age [1, 3]. It has recently been shown that of the remaining half of ALK-negative ALCLs, approximately 20–30% show rearrangements of the DUSP22-IRF4 locus on 6p25.3, 2–8% have rearrangement of TP63 on 3q28 and the remainder lack all three alterations (i.e., “triple negative”), with these alterations appearing to be mutually exclusive in most cases [1, 7, 8]. The DUSP22 gene is immediately telomeric (40 kb) to IRF4 at the 6p25.3 locus, and IRF4/DUSP22 break-apart FISH probes flanking these genes detect rearrangements involving both IRF4 and DUSP22 but cannot distinguish between the two [9, 10]. Rearrangement of this locus is often called DUSP22 rearrangement as it is associated with decreased expression of dual-specificity phosphatase-22, which regulates mitogen-activated protein kinase signaling, but not IRF4 [1, 9, 10]. DUSP22 rearranged ALCLs typically have a favorable prognosis, similar to ALK rearrangements with 5-year overall survival (OS) of 80–90% [1, 4, 11]. Alterations in TP63 are associated with a poor prognosis and lack of any of these rearrangements with an intermediate prognosis, with 5-year OS of 0–17% and 33–42%, respectively [1, 11]. MYC rearrangement has been rarely reported in ALK-negative ALCL with 2 patients recently described by Khanlari et al. both of whom had aggressive disease although one demonstrated a concurrent DUSP22 rearrangement and had longer survival compared to the patient without the alteration [12].
The majority of primary central nervous system lymphomas (PCNSL), non-Hodgkin lymphomas restricted to the CNS, including brain, meninges, spinal cord or eye at presentation, are of the diffuse large B-cell type with T-cell lymphomas constituting less than 5% [13]. ALCLs involving the CNS are very rare, have been ALK-positive, ALK-negative or with unknown ALK status, and tend to exhibit aggressive behavior [5, 13, 14]. Only 15 cases of primary CNS ALK-negative ALCLs have been reported [5, 13, 15,16,17,18,19,20,21,22,23,24]. We describe what may be the first reported case of primary CNS ALK-negative ALCL with DUSP22 rearrangement and review the clinical and pathologic characteristics of CNS ALK-negative ALCLs in the literature.
Clinical summary and pathologic findings
A 55-year-old man with history of hypertension, dyslipidemia, prediabetes, asthma, microcytic anemia likely due to thalassemia, and cardiomyopathy presented with lightheadedness for 2 weeks followed by problems with balance resulting in several falls as well as irritability, memory problems and confusion for 1.5 weeks. He also had difficulty using his right hand. Magnetic resonance imaging (MRI) of the brain showed enlargement and amorphous enhancement involving the left cingulate gyrus, which extended into the left temporal lobe, hippocampus and corpus callosum with extension across the midline (Fig. 1). Additionally, confluent periventricular white matter T2/FLAIR hyperintensity was seen. Cerebrospinal fluid (CSF) cytology performed twice was negative for malignant cells. CSF flow cytometry showed mostly CD5 positive T cells and was interpreted as negative for a lymphoproliferative disorder. HIV testing was negative. Computed tomography (CT) of the neck, chest, abdomen, and pelvis at presentation and repeated approximately one month later showed no convincing evidence of malignancy. Although a PET scan had not been done, CT of the chest, abdomen and pelvis was repeated a third time and was again negative. He also did not have B symptoms of fever, night sweats and weight loss.

Initial axial, contrast-enhanced T1-weighted, magnetic resonance image demonstrating enlargement and amorphous enhancement involving the cingulate gyrus and corpus callosum, extending across the midline (a). There was also confluent periventricular white matter T2/FLAIR hyperintensity (b). Repeat imaging 3 months later showed multiple new contrast-enhancing masses (c) and progression of T2/FLAIR hyperintensity (d)
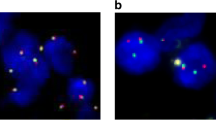
The patient underwent a left frontal brain biopsy that showed atypical lymphoid cells in a background of reactive brain tissue with gliosis and lymphohistiocytic infiltrate. The large cells were positive for CD30 and predominantly negative for CD3. Although features were suspicious for a lymphoproliferative disorder, the sparsity of the atypical cells made definitive diagnosis challenging and also limited molecular testing. He was placed on corticosteroids, but subsequent MRI showed multifocal heterogeneous enhancement with progression of abnormal signals in some regions, with decreasing signals in others. A repeat brain biopsy of the corpus callosum and cingulate gyrus was performed 1.5 months later and showed an infiltrate of many large cells admixed with medium-sized and small lymphocytes in a perivascular and diffuse parenchymal distribution (Fig. 2). The large cells had irregular nuclear contours with small to indistinct nucleoli and moderate amounts of cytoplasm. Scattered hallmark cells with kidney-shaped nuclei were seen, but there were no “doughnut” cells with central nuclear pseudoinclusions or multinucleated cells with “wreath-like” nuclei. Mitoses were easily identified, but no necrosis was seen. The background brain tissue showed gliosis, highlighted by immunohistochemistry for GFAP. The large cells were positive for CD2, CD4, CD25, CD30, and expressed the alpha–beta T-cell receptor (TCR) protein as detected by the antibody βF1 and had negative staining for TCR delta. There was decreased expression of CD3, CD5 and CD7. The neoplastic cells were also positive for granzyme B with a subset positive for TIA-1 and negative for CD8, ALK-1, EMA, clusterin, and EBV EBER. The MIB-1 labeling index was over 90% in the large cells. Less than 5% of the cells were B cells (CD20 +, PAX5 +), and a small population of polytypic plasma cells was present. PCR testing for TCR gene rearrangements showed a clonal gene arrangement at the TRG locus (Fig. 3a). Fluorescence in situ hybridization (FISH) studies were performed with the Vysis FISH ALK break-apart probes against the 5’ to 3’ region of the ALK gene (2p23) and the Leica FISH IRF4/DUSP22 break-apart probes against the 5′ to 3′ region of the IRF4 and DUSP22 genes (6p25.3) which demonstrated an IRF4/DUSP22 gene rearrangement with gain of 3′IRF4 in over 90% of nuclei assessed (Fig. 3b). No ALK gene rearrangement was detected.

Biopsy of the corpus callosum lesion showing large pleomorphic cells, admixed with medium to small lymphocytes, diffusely infiltrating brain parenchyma with perivascular accentuation (arrows) (a, 200 ×). The cells have irregular nuclear contours (b, 400 ×) with scattered hallmark cells (inset 600 ×); mitotic figures are easily seen (arrowheads) (c, 600 ×). On immunohistochemistry, the cells are positive for CD30, with many showing strong membranous and Golgi staining (d, 400 ×); large cells negative for CD30 are likely reactive astrocytes, endothelial cells and/or histiocytes. The tumor cells are also positive for CD2 (e, 400 ×) and granzyme B (f, 400 ×). ALK-1 is negative (g, 400 ×). The MIB-1 labeling index is over 90% (h, 400 ×)

TCRG clonal product (arrows at 245 bp and 218 bp) with two of the primer sets using the BIOMED-2 protocol (a). Fluorescence in situ hybridization demonstrates IRF4/DUSP22 rearrangement (arrows) with an extra copy of 3′IRF4 in the large cells (arrowheads) (b)
Unfortunately, the patient developed seizures and aspiration pneumonia and had to be placed on mechanical ventilation. Repeat brain MRI showed multifocal enhancing masses increased in number and size (Fig. 1c, d). He was placed on multiple anticonvulsant medications and corticosteroids, but due to multiorgan injury could not be given chemotherapy or radiation. He was referred to hospice care and died 2.5 months after the initial surgery.
Discussion
PCNSLs are rare, accounting for 1–2% of non-Hodgkin lymphomas and 2% of all primary brain tumors [13, 14, 25]. The majority are B-cell lymphomas, with DLBCL comprising over 90% [13, 26, 27]. T-cell lymphomas account for approximately 2–4% of all primary CNS lymphomas in the West [28, 29] with a higher incidence in Asia, ranging from 2 to 17% [30,31,32]. Primary CNS T-cell lymphomas (PCNSTL) include peripheral T-cell lymphoma, not otherwise specified, the most common subtype, ALCL, and extranodal NK/T-cell lymphoma [5, 26, 32, 33]. In a series of 45 PCNSTLs from multiple institutions and countries, Shenkier et al. reported a wide age range of presentation from 3 to 84 years with median age of 60 years and male predominance, similar to PCNSLs overall [27, 34]. In a series of primary CNS ALCLs, including ALK-positive, ALK-negative and ALK status unknown, George et al. examined 9 cases, 5 previously reported, and reviewed an additional 4 cases in the literature [5]. All cases evaluated had supratentorial lesions with 2 additionally involving the infratentorial compartment and one also involving the spinal cord [5]. Nine of the 13 cases showed dural/leptomeningeal involvement including 2 cases with sole involvement of the dura, one ALK positive and the other with ALK status unknown [5].
Of the 15 patients with primary CNS ALK-negative ALCL reported in the literature and the current case (Table 1), the median age was 62 (range 22–82). Except for one young female patient who was 22 years old, all patients were above 45 years of age, in contrast to CNS ALK-positive ALCLs, which have been reported in children, similar to systemic ALK-positive ALCL [5, 35]. There was a nearly equal gender distribution with 8 females and 7 males. Nine of 13 patients had a normal immune system. Symptoms spanned a few weeks to several months and ranged from focal neurologic deficits, seizures, and headaches to symptoms of dementia. This highlights the importance of considering CNS lymphoma in the differential diagnosis of atypical dementia symptoms as it is one of the most common diagnoses on biopsies done for neurologic decline of unknown etiology [36]. Nearly all cases involved the supratentorial compartment as solitary or multifocal lesions except in one patient (case 14) who showed leptomeningeal involvement of the posterior fossa and spinal cord [16]. Seven subjects had solitary lesions, whereas 8 had multifocal or diffuse involvement on MRI. Six patients showed leptomeningeal involvement on imaging or histologic examination, some centered in the dura. Seven of 8 patients tested, including the present case, were positive for TCR gene rearrangements with one case resulting in no amplification products (Table 2).
Systemic ALK-positive ALCLs demonstrate several histologic patterns with the “common” pattern of sheet-like growth of hallmark cells with kidney-shaped nuclei, the most frequent pattern among all types of ALCLs [37]. Other variants include the lymphohistiocytic pattern with admixed abundant reactive histiocytes, Hodgkin-like pattern resembling nodular sclerosis classic Hodgkin lymphoma (although most cases previously diagnosed with this pattern are likely classical Hodgkin lymphomas and not ALCLs), and small-cell pattern [3, 4, 37, 38]. Case 3 was reported to be positive for EBV EBER by in situ hybridization and showed abundant histiocytes and eosinophils, suggestive of the Hodgkin-like pattern [5, 19]. As ALCLs are consistently negative for EBV according to the WHO, case 3 may not fit into the category of ALK-negative ALCL with current criteria and may represent rare intracerebral Hodgkin lymphoma [3, 37, 39, 40]. ALK-negative ALCLs show morphologic patterns similar to ALK-positive ALCLs, and a small-cell pattern is not recognized due to overlap with other peripheral T-cell lymphomas [4].
King et al. in a series of systemic ALK-negative ALCLs with DUSP22 rearrangement, have shown that the majority show the common pattern with some cases having slightly smaller cells compared to other genetic subtypes. ALCLs with DUSP22 rearrangement were more likely to show “doughnut” cell morphology with central nuclear pseudoinclusions and less likely to show large pleomorphic and/or multinucleated cells which have been reported to be more common in ALK-negative compared to ALK-positive ALCL but less often seen in ALCL with DUSP22 and TP63 rearrangements [1, 4]. The current case did not demonstrate “doughnut” cells or multinucleated cells. Of the prior reported cases of primary CNS ALK-negative ALCL, no “doughnut” cells were reported, but 5 cases described few to frequent multinucleated cells. Although necrosis was not seen in our case, necrosis is a common feature in PCNSTLs [13] and CNS ALK-negative ALCLs (8 of 15). Perivascular cuffing is also frequently, but not always, seen in primary CNS lymphomas of both B-cell and T-cell lineage, and T-cell lymphomas can be challenging to diagnose as the brain often shows florid reactive changes with gliosis and histiocytic infiltrate, obscuring diffuse parenchymal involvement [13, 14]. On immunohistochemistry, EMA and clusterin are more often negative, and CD2 and CD3 more frequently positive in all genetic subtypes of ALK-negative ALCL compared to ALK-positive ALCL [1, 7]. Similarly, the present case showed absent expression of EMA and clusterin with positivity for CD2 but with decreased expression of CD3. Of the CNS ALK-negative ALCL cases, 9 of 15 had some degree of CD3 immunopositivity, while only 3 of 8 cases on which EMA was performed showed EMA expression (Table 2). While immunohistochemistry for cytotoxic markers is not specific for ALCL subtype, only 5–10% of DUSP22 rearranged cases have been shown to express the cytotoxic markers granzyme B and TIA-1 [1]. The current case was one of the 5 cases positive for TIA-1 and/or granzyme B out of 7 CNS ALK-negative ALCLs on which they were performed. A unique finding in our case is the presence of an extra copy of 3′IRF4 in addition to the IRF4/DUSP22 rearrangement. In a study of 182 patients with cutaneous T-cell lymphoproliferative disorders, Wada et al. found 9 of 45 cutaneous ALCLs and 1 of 32 cases of lymphomatoid papulosis to have an IRF4/DUSP22 translocation. Other IRF4 alterations, most commonly extra copies of IRF4, were seen in a wide variety of T-cell lymphoproliferative disorders, including cutaneous ALCL, and were mutually exclusive with cases harboring IRF4/DUSP22 translocations [10]. The effects of additional cytogenetic alterations on prognosis are unknown; rare cases of DUSP22 rearranged ALCLs demonstrating other alterations, one with concurrent TP63 rearrangement and another with concurrent MYC rearrangement, showed complete remission after chemotherapy although the patient with DUSP22 and MYC rearrangements died 53 months after diagnosis from an unknown cause [8, 12].
There is no consensus on the optimal therapy for PCNSL, but treatment most often consists of high-dose methotrexate as part of multiagent chemotherapy, with or without radiation [41]. Most aggressive systemic lymphomas, including ALCL, are treated with an anthracycline containing chemotherapy regimen, such as cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) [1, 42]; however, these regimens are ineffective in PCNSL in part due to insufficient penetration of the blood–brain barrier (BBB) [41]. Brentuximab vedotin, an antibody–drug conjugate comprised of anti-CD30 monoclonal antibody conjugated to the anti-microtubule agent monomethyl auristatin E, has shown efficacy for relapsed/refractory ALCL but is also not thought to cross the BBB [1, 42, 43].
PCNSLs tend to have a worse prognosis compared to extra-CNS lymphomas [41]. The 5-year OS of PCNSTLs is approximately 30%, similar to that of PCNSL, DLBCL type [44]. ALK-positive ALCL has a better prognosis compared to ALK-negative ALCL also in the CNS, but both appear to have a worse prognosis compared to extra-CNS ALCLs, although studies are limited by the rarity of the disease [5, 35]. Among the CNS ALK-negative ALCLs, 10 of 13 patients with available outcome data died 4 days to 6 months after surgery. Moreover, ALCLs with DUSP22 rearrangement have usually been associated with a good prognosis, but Hapgood et al. reported a 5-year OS of approximately 40% in ALCLs with DUSP22 rearrangement in their series, although their 5-year OS of ALK-positive ALCLs were also lower at 69%, which may be due to the difference in populations studied [7].
In summary, ALK-negative ALCL with DUSP22 rearrangement can also present primarily in the brain and in our case had a poor outcome. T-cell lymphomas are rare in the CNS and, thus, require a high level of suspicion for the appropriate diagnosis to be made. Although not entirely specific, TCR gene rearrangements may be helpful. ALCLs are clinically and genetically heterogeneous, and this case emphasizes the importance of assessing for recurrent fusions involving ALK, DUSP22 and TP63, which have a significant impact on prognosis and management.
References
Parrilla Castellar ER, Jaffe ES, Said JW et al (2014) ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 124:1473–1480
Stein H, Mason DY, Gerdes J et al (1985) The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 66:848–858
Feldman AL, Harris NL, Stein H et al (2017) Anaplastic large cell lymphoma, ALK-negative. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (eds) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. Lyon, IARC, pp 418–421
King RL, Dao LN, McPhail ED et al (2016) Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol 40:36–43
George DH, Scheithauer BW, Aker FV et al (2003) Primary anaplastic large cell lymphoma of the central nervous system: prognostic effect of ALK-1 expression. Am J Surg Pathol 27:487–493
Benharroch D, Meguerian-Bedoyan Z, Lamant L et al (1998) ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 91:2076–2084
Hapgood G, Ben-Neriah S, Mottok A et al (2019) Identification of high-risk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol 186:e28-31
Karube K, Feldman AL (2020) “Double-hit” of DUSP22 and TP63 rearrangements in anaplastic large cell lymphoma, ALK-negative. Blood 135:700
Feldman AL, Dogan A, Smith DI et al (2011) Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 117:915–919
Wada DA, Law ME, Hsi ED et al (2011) Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol 24:596–605
Pedersen MB, Hamilton-Dutoit SJ, Bendix K et al (2017) DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood 130:554–557
Khanlari M, Tang G, Hao S et al (2021) Anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma with MYC rearrangement. Br J Haematol 192:e17-21
Menon MP, Nicolae A, Meeker H et al (2015) Primary CNS T-cell lymphomas: a clinical, morphologic, immunophenotypic, and molecular analysis. Am J Surg Pathol 39:1719–1729
Giannini C, Dogan A, Salomao DR (2014) CNS lymphoma: a practical diagnostic approach. J Neuropathol Exp Neurol 73:478–494
Sugino T, Mikami T, Akiyama Y et al (2013) Primary central nervous system anaplastic large-cell lymphoma mimicking lymphomatosis cerebri. Brain Tumor Pathol 30:61–65
Lannon M, Lu JQ, Chum M et al (2020) ALK-negative CNS anaplastic large cell lymphoma: case report and review of literature. Br J Neurosurg. https://doi.org/10.1080/02688697.2020.1839630
Paulus W, Ott MM, Strik H et al (1994) Large cell anaplastic (Ki-1) brain lymphoma of T-cell genotype. Hum Pathol 25:1253–1256
Nuckols JD, Liu K, Burchette JL et al (1999) Primary central nervous system lymphomas: a 30-year experience at a single institution. Mod Pathol 12:1167–1173
Chuang SS, Huang W, Lin CN et al (2001) Primary cerebral anaplastic large cell lymphoma containing abundant reactive histiocytes and eosinophils. A case report and literature review. Pathol Res Pract 197:647–652
Gonzales M (2003) Primary meningeal anaplastic large cell lymphoma. Pathology 35:451–452
Tajima Y, Miyazaki Y, Higashi T et al (2003) Primary CD30/Ki-1 positive anaplastic large cell lymphoma of the central nervous system occurring in a patient with a seventeen-year history of essential thrombocythemia. Leuk Lymphoma 44:1243–1245
Rowsell EH, Zekry N, Liwnicz BH et al (2004) Primary anaplastic lymphoma kinase—negative anaplastic large cell lymphoma of the brain in a patient with acquired immunodeficiency syndrome. Arch Pathol Lab Med 128:324–327
Kodama K, Hokama M, Kawaguchi K et al (2009) Primary ALK-1-negative anaplastic large cell lymphoma of the brain: case report and review of the literature: case report. Neuropathology 29:166–171
Colen CB, Rayes M, Kupsky WJ et al (2010) Synchronous meningioma and anaplastic large cell lymphoma. Neuropathology 30:260–266
Ostrom QT, Patil N, Cioffi G et al (2020) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol 22:iv1-96
Lim T, Kim SJ, Kim K et al (2011) Primary CNS lymphoma other than DLBCL: a descriptive analysis of clinical features and treatment outcomes. Ann Hematol 90:1391–1398
Rubenstein J, Ferreri AJM, Pittaluga S (2008) Primary lymphoma of the central nervous system: epidemiology, pathology and current approaches to diagnosis, prognosis and treatment. Leuk Lymphoma 49:43–51
Ferreri AJM, Blay JY, Reni M et al (2003) Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol 21:266–272
Bataille B, Delwail V, Menet E et al (2000) Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg 92:261–266
Choi JS, Nam DH, Ko YH et al (2003) Primary central nervous system lymphoma in Korea: comparison of B- and T-cell lymphomas. Am J Surg Pathol 27:919–928
Shibamoto Y, Ogino H, Suzuki G et al (2008) Primary central nervous system lymphoma in Japan: changes in clinical features, treatment, and prognosis during 1985–2004. Neuro Oncol 10:560–568
Pang Y, Chihara D (2021) Primary and secondary central nervous system mature T- and NK-cell lymphomas. Semin Hematol 58:123–129
Miyata-Takata T, Takata K, Kato S et al (2017) Clinicopathological analysis of primary central nervous system NK/T cell lymphoma: rare and localized aggressive tumour among extranasal NK/T cell tumours. Histopathology 71:287–295
Shenkier TN, Blay JY, O’Neill BP et al (2005) Primary CNS lymphoma of T-cell origin: a descriptive analysis from the International Primary CNS Lymphoma Collaborative Group. J Clin Oncol 23:2233–2239
Nomura M, Narita Y, Miyakita Y et al (2013) Clinical presentation of anaplastic large-cell lymphoma in the central nervous system. Mol Clin Oncol 1:655–660
Magaki S, Gardner T, Khanlou N et al (2015) Brain biopsy in neurologic decline of unknown etiology. Hum Pathol 46:499–506
Jaffe ES (2001) Anaplastic large cell lymphoma: the shifting sands of diagnostic hematopathology. Mod Pathol 14:219–228
Kinney MC, Collins RD, Greer JP et al (1993) A small-cell-predominant variant of primary Ki-1 (CD30)+ T-cell lymphoma. Am J Surg Pathol 17:859–868
Herling M, Rassidakis GZ, Jones D et al (2004) Absence of Epstein-Barr Virus in anaplastic large cell lymphoma: a study of 64 cases classified according to World Health Organization criteria. Hum Pathol 35:455–459
Johnson MD, Kinney MC, Scheithauer BW et al (2000) Primary intracerebral Hodgkin’s disease mimicking meningioma: case report. Neurosurgery 47:454–457
Grommes C, DeAngelis LM (2017) Primary CNS lymphoma. J Clin Oncol 35:2410–2418
Hapgood G, Savage KJ (2015) The biology and management of systemic anaplastic large cell lymphoma. Blood 126:17–25
Pierce JMR, Mehta A (2017) Diagnostic, prognostic and therapeutic role of CD30 in lymphoma. Expert Rev Hematol 10:29–37
Chihara D, Fowler NH, Oki Y et al (2018) Impact of histologic subtypes and treatment modality among patients with primary central nervous system lymphoma: a SEER database analysis. Oncotarget 9:28897–28902
Acknowledgements
The authors would like to thank Drs. Niroshi Senaratne, Thomas Lee and Wayne Grody at UCLA Medical Center, Los Angeles, California, for their expertise in cytogenetics and molecular pathology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Magaki, S., Satyadev, R., Chen, Z. et al. Central nervous system ALK-negative anaplastic large cell lymphoma with IRF4/DUSP22 rearrangement. Brain Tumor Pathol 39, 25–34 (2022). https://doi.org/10.1007/s10014-021-00415-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-021-00415-0