
Abstract
Activin receptor-like kinase-1 (ALK-1), the gene mutated in HHT type 2 (HHT2), is a serine/threonine kinase receptor type I of the TGF-β superfamily, specifically expressed on endothelial cells. We established an HHT2 genotype in 16 families and report nine novel mutations. These include insertions and deletions of single base pairs in exons 3, 8 and 9, as well as nonsense mutations in exons 4 and 8 of ALK-1, which would lead to premature truncation and unstable mRNA or protein. Three novel missense mutations were identified in exons 7 and 8 of the kinase domain. Five previously reported substitutions were also observed in the families analyzed. Our results bring to 36, the number of mutations associated with HHT2, and are mostly found in exons 8 and 3 followed by exons 4 and 7. To ascertain the potential functional implications of the missense mutations in the ALK-1 kinase domain, we generated a model based on the three-dimensional structure of the homologous ALK-5 kinase domain. Our data reveal that the 11 missense mutations modify residues conserved among type I receptors and alter the polarity, charge, hydrophobicity and/or size of the substituted amino-acid and likely lead to misfolded and nonfunctional proteins.
Similar content being viewed by others

Introduction
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder characterized by epistaxis (recurrent nosebleeds), mucocutaneous telangiectases, and arteriovenous malformations (AVMs) in lung, brain, and liver. Endoglin (ENG) and ALK-1 are the genes mutated in HHT type 1 (HHT1) and HHT type 2 (HHT2), respectively. Mutations in these genes lead to similar phenotypes, although HHT1 is associated with a higher incidence of pulmonary AVMs while HHT2 tends to be milder and has a later onset.1,2,3
To date, 55 distinct ENG mutations and 27 ALK-1 mutations have been reported. Protein expression studies, in human umbilical vein endothelial cells (HUVECs) and peripheral blood activated monocytes, have confirmed haploinsufficiency as the prevalent model for HHT1 since mutant proteins are generally not expressed.4,5,6,7 Reduced ALK-1 levels were also observed on HUVECs of newborns with HHT2, suggesting that mutant ALK-1 proteins might also be nonfunctional.8
Like other type I receptors, ALK-1 consists of an N-terminal extracellular domain with 10 conserved Cys residues, a transmembrane region, and an intracellular region consisting almost entirely of serine/threonine kinase domains.9 ALK-1 shares a relatively high amino-acid identity in the kinase domain to ALK-2 (77%), ALK-5 (63%), ALK-4 (59%) and ALK-3 (58%).10,11
In this study we performed molecular analysis in families with a clinical diagnosis of HHT and report nine novel mutations in the ALK-1 gene. Using homology modeling and the three-dimensional structure of ALK-5,12,13 we generated a structure of the ALK-1 kinase domain and discuss possible structural and functional consequences of the known missense mutations on this domain.
Materials and methods
Patients and samples
A clinical diagnosis of HHT is based on the presence of three of four criteria: epistaxis, telangiectases, a visceral manifestation (AVM or gastrointestinal bleeds), and a family history of HHT.14 Thus, clinically affected individuals with normal endoglin levels relative to age-matched controls8 (and data not shown) are considered putative HHT2 candidates and their DNA samples are first analyzed for ALK-1 mutations.
Blood samples and medical histories were obtained with informed consent from all individuals participating in the study. For newborns, consent was obtained from parents and umbilical cord and placenta samples prepared as described previously.4,6 All procedures were reviewed and approved by the Research Ethics Board of the Research Institute of the Hospital for Sick Children, Toronto, Canada.
Mutation analysis
Genomic DNA was prepared from peripheral blood lymphocytes, placenta and HUVEC using Puregene® DNA Isolation Kit (Gentra Systems) according to the manufacturer's protocols.
All exons of ALK-1 were analyzed using the Open Gene Automated DNA Sequencing System II (VGI).6,8 Two sets of primers were used for amplification and sequencing of each of the nine coding exons and were designed to cover exon/intron boundaries allowing for detection of alternative splice sites and flanking intronic polymorphisms.
ALK-1 modeling
Alignments of human ALKs were made using PILEUP from Genetics Computer Group (GCG) package, exported as multiple sequence files (MSF) and displayed using the program BOXSHADE. The crystal structure of the kinase domain of ALK-512,13 was used to build a model of the ALK-1 kinase domain using homology modeling techniques.15 Using SwissPDB program, amino-acid residues of the ALK-5 kinase domain (PDBID: 1b6c) were substituted with the respective amino-acids of ALK-1 to generate a homology model of the (wild type) ALK-1 kinase domain. To examine the possible effects on this model of selected missense mutations, amino-acid residue side chains were altered to correspond to the known ALK-1 missense mutations.
Results
Mutation analysis in 16 families with HHT2
A genotype of HHT2 was established in 16 families. We identified 14 ALK-1 mutations, including nine novel ones (Figure 1 and Table 1). They were single base pair deletions, insertions and substitutions distributed in exons 3 and 4, corresponding to extracellular and transmembrane regions, respectively, and in exons 7, 8 and 9 of the intracellular kinase domain.

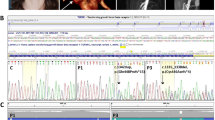
Nine novel mutations identified in HHT2 patients. ALK-1 exons amplified from genomic DNA were sequenced using the Microgeneblaster® automated sequencer (VGI). (a) In exon 3, deletion of G86 and insertion of G238 are shown with respective controls. (b) C430T and G986A substitutions present in exons 4 and 7. (c) In exon 8, G1121A and T1123C substitutions were found in adjacent codons and a G1113 insertion was identified. (d) G1171T substitution and C1299 deletion were found in exons 8 and 9, respectively. (Base codes: M=A or C; S=G or C; Y=C or T; K=G or T; R=A or G).
Mutations in the extracellular and transmembrane domains
We identified two novel mutations in exon 3. In family 38, a G86 deletion causing a frameshift at codon 29, was found in DNA from proband H118 (Figure 1a and Table 1). This patient had epistaxis, telangiectases, and a liver AVM. Four clinically affected adults and a 12-year-old boy who had not yet manifested signs of HHT, also carried the familial mutation while two nonaffected relatives did not. The second novel mutation was an insertion of G at position 238 creating a frameshift at codon 80 (Figure 1a and Table 1). It was identified in a clinically affected woman (H449) of family 133, who had a pulmonary AVM, but not in two unaffected relatives.
Two mutations were identified in exon 4. A previously reported nonsense mutation, G423A substitution,16 converting Trp141 into a stop was detected in the proband H762 of family 198. This patient showed severe manifestations of HHT including hepatic AVM and gastrointestinal bleeds. The mutation was not detected in two nonaffected family members (Table 1). A novel C430T substitution, causing a stop at Arg144 (Figure 1b and Table 1) was found in proband H445 of family 131 and her newborn daughter. The proband's sister was also clinically affected but her newborn son did not carry the familial mutation.
Mutations in the intracellular kinase domain
In all, 10 mutations were identified in the ALK-1 kinase domain. Two missense mutations were detected in exon 7. A novel G986A substitution that mutates Arg329 to His was found in proband H180 of family 59, who had both pulmonary and cerebral AVMs (Figure 1b and Table 1), and in one of her two young children. A G998T substitution, converting Ser333 to Ile, was found in four clinically affected individuals of family 41 (Table 1). This mutation was reported previously in a large family geographically linked to the current one, suggesting a common ancestor.16,17
Exon 8 harbored half of the mutations (7/14) identified in this study (Table 1). Four of these are novel: one insertion, two missense and one nonsense mutations. The insertion of G1113, causing a frameshift at residue 372, was identified in proband H334 of family 106 (Figure 1c and Table 1). If translated, this protein would lack part of the kinase domain and likely be nonfunctional. The mutation was present in eight of 15 family members tested and correlated with clinical manifestations.

Nine novel mutations identified in HHT2 patients. ALK-1 exons amplified from genomic DNA were sequenced using the Microgeneblaster® automated sequencer (VGI). (a) In exon 3, deletion of G86 and insertion of G238 are shown with respective controls. (b) C430T and G986A substitutions present in exons 4 and 7. (c) In exon 8, G1121A and T1123C substitutions were found in adjacent codons and a G1113 insertion was identified. (d) G1171T substitution and C1299 deletion were found in exons 8 and 9, respectively. (Base codes: M=A or C; S=G or C; Y=C or T; K=G or T; R=A or G).
Three of the missense mutations present in exon 8 altered two adjacent amino-acids (Table 1). A previously reported C1120T substitution16,18 was found in two unrelated families and converts Arg374 into Trp. In family 82, the mutation was found in proband H271 with pulmonary AVM and an affected sibling, but not in a nonaffected relative. In family 167, three of nine patients tested exhibited clinical signs of HHT, which correlated with the presence of the mutation. The substitution G1121A was found in family 129 (Figure 1c) and changes Arg374 to Gln. This mutation was found in proband H434 who had gastrointestinal bleeds and in an affected sibling. Another novel mutation, T1123C, that converts Tyr375 to His, was identified in family 75 (Figure 1c). It was found in proband H260 with pulmonary AVM and two clinically affected individuals as well as in a newborn.
We report the first nonsense mutation in exon 8, a G1171T substitution detected in family 157 (Figure 1d). It leads to a termination codon at Glu391, and the translated protein would likely be inactive due to partial loss of the kinase domain. This mutation was detected in three clinically affected individuals but not in a relative without signs of HHT.
Another two missense mutations affecting the same amino-acid, were identified (Table 1). A recently published substitution, C1231T, that changes Arg411 to Trp19 was found in unrelated family 170. This mutation was present in four clinically affected individuals, including proband H607 with a pulmonary AVM, and absent from seven nonaffected relatives. The second mutation, a G1232A substitution that converts Arg411 into Gln was previously reported in three studies.16,20,21 We found this mutation in two seemingly unrelated families. In family 20, it was detected in two clinically affected individuals, while in family 185 it was found in five individuals with clinical signs and absent from four without symptoms.
A novel mutation was identified in exon 9: deletion of C1299 in family 181 (Figure 1d) that creates a frameshift at amino-acid 434. It was present in two of four samples tested and correlated with HHT signs.
The newly described mutations bring to 36, the total number of known mutations in the ALK-1 gene (Figure 2).

(a) An alignment of ALK-1 (residues 183–503) and ALK-5 (residues 186–503) cytoplasmic kinase domains. Identical amino-acids are represented by dark boxes while conservative substitutions are shown as gray boxes. The position of the 13 known ALK-1 missense mutations is indicated above the alignment. The GS domain and the 11 kinase subdomains are shown below the alignment. (b) Summary of known mutations in the ALK-1 gene. A map of the ALK-1 gene with the 36 published mutations is illustrated approximately to scale. The coding region (exons 2–10) is represented as open boxes and noncoding exons as gray boxes.
Polymorphisms in the ALK-1 gene
Four of the substitutions observed, including three intronic, were considered neutral polymorphims as they were found in normal alleles. Substitution IVS1-38C>T was found in 14 of 100 individuals tested. Substitution IVS3+11C>T was expressed as a single allele in 40% and on both alleles in 6% of 159 individuals tested. Substitution IVS9-30G>A was found in 2 of 100 individuals tested (2%). The substitution G747A was also found in 2 of 100 individuals tested; this is a silent mutation that does not alter Val 249.
Homology modeling of ALK-1
To compare ALK-1 and ALK-5 structures, we first aligned their amino-acid sequences, revealing the overall similarity of the GS and kinase domains (Figure 2a). The 11 mutated amino-acids, representing 13 known missense mutations in the ALK-1 kinase domain all correspond to conserved residues. When compared to other TGF-β superfamily type I receptors (GenBank sequences at http://www.ncbi.nlm.nih.gov/), 10 of these residues were identical in the six mammalian type I receptors (ALK-1 to ALK-6) while the 11th (Ile398) differed only in ALK-6 where it was substituted with Met. All 11 normal amino-acids were also conserved in mouse and rat.
A structural model of the cytoplasmic domain of ALK-1 was generated by computer modeling based on the homologous structure of ALK-5. The catalytic domain of ALK-1 is predicted to contain a small N-lobe dominated by a five-stranded β-sheet and a larger, mostly helical C-lobe (Figure 3). The 11 mutated amino-acids were localized onto this molecular model and found to be concentrated in the core of the kinase C-lobe. One of the 11 mutated side chains (Cys344) was contained in a β-pleated sheet, three (Ile398, Glu407, Arg484) were within α-helical structures, while the remaining seven were in the loops between β-pleated sheets (Arg329, Ser333) and/or α-helices (Arg374, Tyr375, Met376, Arg411, Pro424) (Figure 3).

Effects of missense mutations on the structure of ALK-1
Each of the 13 missense mutations was then analyzed for its position in the kinase C-lobe. Potential alterations, if any, in the hydrogen bonding scheme, and/or presence of induced steric clashes, were considered. For example, Arg329 and Ser333 are conserved residues of the catalytic segment in subdomain VI of type I receptors and are part of one of the two consensus motifs that confer specificity in either serine/threonine or tyrosine kinases.9,22 This motif, His328–Arg–Asp–Leu–Lys–X–X–Asn335, contains two invariant residues – Asp330 and Asn335 – that are implicated in ATP binding and phosphotransfer,9 while Glu334 is involved in substrate binding by forming an ion pair with Arg329 in the consensus sequence. Upon mutating Arg329 to His, a basic residue is replaced with a structurally distinct residue with a side chain imidazole ring. Modeling suggests this could lead to the formation of a new hydrogen bond to the backbone of Val 353 of the β9-pleated sheet (Figure 4a, c) and thus potentially interfere with substrate binding. Ser333Ile replaces a purely hydrocarbon side chain with a relatively hydrophilic side chain. Both mutated residues, located in the catalytic loop between the E-helix and the activation segment, could disrupt the activation site and interfere with catalysis.

Examples of potential alterations in ALK-1 structure resulting from missense mutations, as suggested by molecular modeling. (a, c) An Arg329His substitution removes a weak H-bond between Arg329 and Asn366 and can produce instead a strong H-bond between His329 and Val353 backbone. (b, d) With Arg374Trp substitution, the structural stability gained from wild-type Arg374 side chain H-bonding scheme to backbone substituents of Phe425, Met438 and Val439, is lost. The Trp374 indole ring may interact instead with substituents on Asp432 and Pro433. Wild type and corresponding mutated residues are shown in red. Strong H-bonding is shown in green, weak H-bonding in gray, and possible steric clashes induced by mutations are rendered as purple arrows. Hydrogen-bonding schemes (wild type and mutants) are as calculated by the SwissPDB program.
Four missense mutations affect three consecutive amino-acids (Arg374, Tyr375, and Met376) found in the loop preceding the αEF helix. They fall within subdomain VIII containing an Ala–Pro–Glu motif, which is highly conserved among protein kinases. The eight amino-acids preceding this sequence are conserved and form part of a consensus (Gly371–Thr/Ser–X–X–Tyr/Phe–X–Ala–Pro–Glu) specific to serine/threonine kinases.9,22,23 Subdomain VIII plays an important role in substrate recognition by providing a pocket to accommodate hydrophobic residues. Two mutations (Gln and Trp) affect Arg374; the possible participation of Trp374 in alternative H-bonding schemes versus wild-type Arg374 is illustrated in Figure 4b,d.
Arg484, located in the αI helix, is part of a highly conserved COOH-terminal sequence between residues 479 and 489 of ALK-1. This region, referred to as NANDOR BOX (for non-activating non-down-regulating), is highly conserved among type I receptors in mammals, Drosophila and Xenopus and was shown to be critical for ligand-induced TGFβ-receptor signaling and downregulation.24 ALK-5 missense mutations in NANDOR BOX resulted in the inability of TGF-β to stimulate expression of PAI-1, fibronectin, and Smad2 phosphorylation.25,26 An Arg484Trp mutation that falls within this motif would clearly be implicated in a loss of function.
Discussion
We described nine novel ALK-1 mutations, bringing to 36 the total number of known mutations in HHT2 (Figure 2b). Nine (25%) of these are present in exon 3, ten (28%) in exon 8, five in each of exons 4 and 7, two in each of exons 6, 9, 10 and one in exon 2. These results suggest that sequencing exons 8, 3, 4 and 7 of ALK-1 should be performed first to accelerate the process of mutation identification in HHT2.
The majority (70%) of HHT2-associated ALK-1 mutations are single base-pair substitutions leading to missense and nonsense mutations. In contrast, most (65%) HHT1-associated ENG mutations cause frameshifts and premature stop codons. Furthermore, large insertions, deletions and splice site mutations are quite common in HHT1 but so far have not been identified in HHT2 (Cymerman et al6 and Paquet et al7 and data not shown). This might be partially due to the almost exclusive use of direct sequencing in ALK-1 mutation analysis. In our own series, we have now resolved 59 HHT1 families using quantitative multiplex PCR (QMPCR) and sequencing and 22 HHT2 families using mostly sequencing. We will now perform ALK-1 QMPCR on probands with a clinical diagnosis of HHT and normal levels of endoglin protein, assumed to be potential HHT2 candidates. Although AVMs are presumed more frequent in HHT1 than HHT2,1,2,3 it is not possible to determine the HHT type clinically. As seen in Table 1, seven of the 16 probands had an AVM: two – hepatic, four – pulmonary, and one – both pulmonary and cerebral. This represents the highest incidence of AVMs reported in a series of HHT2 families although previous studies have reported AVMs in two large HHT2 kindreds.17,18
Mutations are distributed throughout the ALK-1 gene. Frameshift and nonsense mutations predict nonsense mediated mRNA decay27 and failure to produce functional protein. The missense mutations reported generally alter conserved amino-acids and likely result in structural changes. Six of the 19 ALK-1 missense mutations were found in exon 3 of the extracellular domain. We have previously demonstrated that HUVECs with Gly48Glu/Ala49Pro and Trp50Cys mutations expressed reduced levels of ALK-1.8 In addition, introduction of the Trp50Cys mutation into the extracellular domain of the ALK-1/ALK-5 chimera led to the abrogation of signaling activity due to low levels of expression of the unstable chimeric receptor.28 The structural alterations caused by these missense mutations likely result in protein misfolding and intracellular degradation explaining the lack of surface expression of mutant proteins. A site for possible interaction between R-I and R-II receptors was identified in the extracellular domain of ALK-5.29 Several ALK-1 residues affected by missense mutations align with a structurally conserved region important for ALK-5 signaling and ligand internalization, but not ligand binding. Since ALK-1 can function as a receptor for TGF-β in endothelial cells,30 we can speculate that certain mutations in this extracellular region might impair R-I/R-II interactions, ligand-dependent signaling and/or ligand internalization.
The missense mutations found in the kinase domain occur at residues that are conserved among type I receptors. Homology modeling and analysis of the missense mutations within the ALK-1 kinase domain shows that they alter the polarity, charge, hydrophobicity, and/or size of the substituted amino-acid and will likely have structural effects creating misfolded and unstable proteins. Such nonfunctional mutant proteins would support the haploinsufficiency model. However, we cannot rule out the possibility that some of the missense mutations lead to dominant-negative effects; engineering of selected mutants should clarify this point.
TGF-β is thought to regulate endothelial function via a balance between ALK-1 and ALK-5 signaling.30 ALK-5 effects mediated by Smad2/3 lead to stimulation of extracellular matrix production and vessel maturation, while signaling through ALK-1 and Smad1/5 stimulates proliferation and migration of endothelial cells.31 Endoglin is thought to play a key role in the regulation of the ALK-1/ALK-5 balance, which is critical in vascular remodeling. This might explain why ENG and ALK-1 mutations lead to a similar vascular pathology.
Mutations in BMPRII or ALK-1 genes can lead to a distinct disorder, primary pulmonary hypertension.19,32,33 Since both receptors act through Smad1/5, one can speculate that this pathway is particularly important for endothelial function. Our analysis of missense mutations in the kinase domain of ALK-1 using homology modeling, further emphasizes the importance of the TGF-β receptor family in sustaining normal vascular structure and function. Determining the mode of action of ALK-1 is essential to our understanding of the mechanisms responsible for maintenance of vascular integrity, as disruption of its function generally leads to HHT but can also cause pulmonary hypertension.
References
Heutink P, Haitjema T, Breedveld GJ et al: Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity. J Med Genet 1994; 31: 933–936.
McAllister KA, Lennon F, Bowles-Biesecker B et al: Genetic heterogeneity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31: 927–932.
Berg JN, Guttmacher AE, Marchuk DA, Porteous ME : Clinical heterogeneity in hereditary haemorrhagic telangiectasia: are pulmonary arteriovenous malformations more common in families linked to endoglin? J Med Genet 1996; 33: 256–257.
Pece N, Vera S, Cymerman U, White RI, Wrana JL, Letarte M : Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest 1997; 100: 2568–2579.
Pece-Barbara N, Cymerman U, Vera S, Marchuk DA, Letarte M : Expression analysis of four endoglin missense mutations suggests haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum Mol Genet 1999; 8: 2171–2181.
Cymerman U, Vera S, Pece-Barbara N et al: Identification of hereditary hemorrhagic telangiectasia type 1 in newborns by protein expression and mutation analysis of endoglin. Pediatr Res 2000; 47: 24–35.
Paquet ME, Pece-Barbara N, Vera S et al: Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum Mol Genet 2001; 10: 1347–1357.
Abdalla SA, Pece-Barbara N, Vera S et al: Analysis of ALK-1 and endoglin in newborns from families with hereditary hemorrhagic telangiectasia type 2. Hum Mol Genet 2000; 9: 1227–1237.
Hanks SK, Quinn AM, Hunter T : The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 1988; 241: 42–52.
ten Dijke P, Ichijo H, Franzen P et al: Activin receptor-like kinases: a novel subclass of cell surface receptors with predicted serine/threonine kinase activity. Oncogene 1993; 8: 2879–2887.
Kingsley DM : The TGF-β superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev 1994; 8: 133–146.
Huse M, Chen YG, Massague J, Kuriyan J : Crystal structure of the cytoplasmic domain of the type I TGF β receptor in complex with FKBP12. Cell 1999; 96: 425–436.
Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massague J : The TGF β receptor activation process: an inhibitor- to substrate-binding switch. Mol Cell 2001; 8: 671–682.
Shovlin C, Guttmacher A, Buscarini E et al: Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu–Osler–Weber Syndrome). Am J Med Genet 2000; 91: 66–67.
Combet C, Jambon M, Deleage G, Geourjon C : Geno 3D: automatic comparative molecular modelling of prrotein. Bioinformatics 2002; 18: 213–214.
Berg JN, Gallione CJ, Stenzel TT et al: The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61: 60–67.
McDonald JE, Miller FJ, Hallam SE, Nelson L, Marchuk DA, Ward KJ : Clinical manifestations in a large hereditary hemorrhagic telangiectasia (HHT) type 2 kindred. Am J Med Genet 2000; 93: 320–327.
Kjeldsen AD, Brusgaard K, Poulsen L et al: Mutations in the ALK-1 gene and the phenotype of hereditary hemorrhagic telangiectasia in two large Danish families. Am J Med Genet 2001; 98: 298–302.
Trembath RC, Thomson JR, Machado RD et al: Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345: 325–334.
Johnson DW, Berg JN, Baldwin MA et al: Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13: 189–195.
Lin WD, Wu JY, Hsu HB, Tsai FJ, Lee CC, Tsai CH : Mutation analysis of a family with hereditary hemorrhagic telangiectasia associated with hepatic arteriovenous malformation. J Formos Med Assoc 2001; 100: 817–819.
Hanks SK, Quinn AM : Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol 1991; 200: 38–62.
Hanks SK, Hunter T : Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J 1995; 9: 576–596.
Garamszegi N, Dore JJ, Penheiter SG, Edens M, Yao D, Leof EB : Transforming growth factor beta receptor signaling and endocytosis are linked through a COOH terminal activation motif in the type I receptor. Mol Biol Cell 2001; 12: 2881–2893.
Hocevar BA, Brown TL, Howe PH : TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J 1999; 18: 1345–1356.
Engel ME, McDonnell MA, Law BK, Moses HL : Interdependent SMAD and JNK signaling in transforming growth factor-β-mediated transcription. J Biol Chem 1999; 274: 37413–37420.
Maquat LE, Carmichael GG : Quality control of mRNA function. Cell 2001; 104: 173–176.
Lux A, Attisano L, Marchuk DA : Assignment of transforming growth factor beta1 and beta3 and a third new ligand to the type I receptor ALK-1. J Biol Chem 1999; 274: 9984–9992.
Guimond A, Sulea T, Zwaagstra JC, Ekiel I, O'Connor-McCourt MD : Identification of a functional site on the type I TGF-β receptor by mutational analysis of its ectodomain. FEBS Lett 2002; 513: 147–152.
Oh P, Seki T, Goss KA et al: Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 2000; 97: 2626–2631.
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P : Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J 2002; 21: 1743–1753.
Deng Z, Morse JH, Slager SL et al: Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67: 737–744.
Newman JH, Wheeler L, Lane KB et al: Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 2001; 345: 319–324.
Acknowledgements
We are grateful to all the patients and family members who participated in the study and to the genetic counselors and physicians for providing the patient information. We thank Sonia Vera for protein analysis and preparation of some DNA samples. This work was supported by grant HHT-FY01-554 from March of Dimes and grant T 5016 from the Heart and Stroke Foundation of Ontario (ML).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdalla, S., Cymerman, U., Johnson, R. et al. Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur J Hum Genet 11, 279–287 (2003). https://doi.org/10.1038/sj.ejhg.5200919
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200919
Keywords
This article is cited by
-
Structural basis for ALK2/BMPR2 receptor complex signaling through kinase domain oligomerization
Nature Communications (2021)
-
The clinical and genetic features of hereditary haemorrhagic telangiectasia (HHT) in central South Africa—three novel pathogenic variants
Molecular Biology Reports (2020)
-
Hereditary hemorrhagic telangiectasia in Japanese patients
Journal of Human Genetics (2014)
-
Retention in the endoplasmic reticulum is the underlying mechanism of some hereditary haemorrhagic telangiectasia type 2 ALK1 missense mutations
Molecular and Cellular Biochemistry (2013)
-
Mutation analysis of "Endoglin" and "Activin receptor-like kinase" genes in German patients with hereditary hemorrhagic telangiectasia and the value of rapid genotyping using an allele-specific PCR-technique
BMC Medical Genetics (2009)
