
Abstract
Fifteen years ago, a role for excitotoxic damage in the pathology of amyotrophic lateral sclerosis (ALS) was postulated. This stimulated the development of riluzole, the only available treatment for the disease. Since then, the identification of abnormal forms of Superoxide dismutase as the genetic basis of certain familial forms of ALS has provided a huge impetus to the search for new effective treatments for this devastating disease. Transgenic mouse models have been developed expressing these aberrant mutants that develop a form of motor neurone disease the progress of which can be slowed by riluzole. Studies in these mice have provided evidence for a role for excitotoxic, apoptotic and oxidative processes in the development of pathology. The mice can be used for testing molecules targeting these processes as potential therapies, to allow the most promising to be evaluated in humans. Several such agents are currently in clinical trials.
Many previous clinical trials in ALS were insufficiently powered to demonstrate any relevant effect on disease progression. This situation has been to some extent remedied in the more recent trials, which have recruited many hundreds of patients. However, with the exception of studies with riluzole, the results of these have been disappointing. In particular, a number of large trials with neurotrophic agents have revealed no evidence for efficacy.
Nonetheless, the need for large multinational trials of long duration limits the number that can be carried out and makes important demands on investment. For this reason, surrogate markers that can be used for rapid screening in patients of potential treatments identified in the transgenic mice are urgently needed.


Similar content being viewed by others

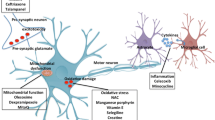

Notes
Use of tradenames is for product identification only and does not imply endorsement.
References
Havercamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population: validation of a scoring system and a model for survival prediction. Brain 1995; 118: 707–19
Swash M. Clinical features and diagnosis of amyotrophic lateral sclerosis. In: Brown Jr RH, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz; 2000: 3–30
Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn Superoxide dismutasegene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362: 59–62
Deng H-X, Hentati A, Tainer JA, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn Superoxide dismutase. Science 1993; 261: 1047–51
Cudkowicz ME, McKenna-Yasek D, Sapp PE, et al. Epidemiology of mutations in Superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol 1997; 41: 210–21
Gurney ME, Liu R, Althaus JS, et al. Mutant CuZn Superoxide dismutase in motor neuron disease. J Inherit Metab Dis 1998; 21: 587–97
Bensimon G, Lacomblez L, Meininger V, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med 1994; 330: 585–91
Lacomblez L, Bensimon G, Leigh PN, et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996; 347: 1425–31
Meininger V. Efficacy of riluzole in the treatment of amyotrophic lateral sclerosis. Rev Contemp Pharmacol 1997; 8: 255–64
Doble A. Effects of riluzole on glutaminergic neurotransmission in the mammalian central nervous system and other pharmacological effects. Rev Contemp Pharmacol 1997; 8: 213–26
Miller R, Mitchell J, Lyon M, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Available in The Cochrane Library [database on disk and CD ROM]. Updated quarterly. The Cochrane Collaboration; issue 2. Oxford: Update Software, 2002: CD001447
Gurney ME, Pu H, Chiu AY. Motor neuron degeneration in mice that express a human Cu, Zn Superoxide dismutase mutation. Science 1994; 264: 1772–5
Price DL, Sisodia SS, Borchelt DR. Genetic neurodegenerative diseases: the human illness and transgenic models. Science 1998; 282: 1079–83
Green SL, Tolwani RJ. Animal models for motor neuron disease. Lab Anim Sci 1999; 49: 480–7
Nagai M, Aoki M, Miyoshi I, et al. Rats expressing human cytosolic copper-zinc Superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci 2001; 21: 9246–54
Doble A, Kennel P. Animal models of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 301–12
Gurney ME. Transgenic animal models of amyotrophic lateral sclerosis. In: Brown Jr RH, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz, 2000: 251–62
Shibata N. Transgenic mouse model for familial amyotrophic lateral sclerosis with Superoxide dismutase-1 mutation. Neuropathology 2001; 21: 82–92
Dal Canto MC, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu,Zn Superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 1997; 93: 537–50
Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of amyotrophic lateral sclerosis. Am J Pathol 1994; 145: 1271–9
Dal Canto MC, Gurney ME. Neuropathological changes in 2 lines of mice carrying a transgene for mutant human Cu, Zn, SOD and in mice overexpressing wild-type human SOD-a model of familial amyotrophic lateral sclerosis (FALS). Brain Res 1995; 676: 25–40
Ikonomidou C, Qin Qin Y, Labruyere J, et al. Motor neuron dehgeneration induced by excitotoxin agonists has features in common with those seen in the SOD-1 transgenic mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 1996; 55: 211–24
Tu PH, Raju P, Robinson KA, et al. Transgenic mice carrying a human mutant Superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 1996; 93: 3155–60
Kato S, Nakashima K, Horiuchi S, et al. Formation of advanced glycation end-product-modified Superoxide dismutase-1 (SOD1) is one of the mechanisms responsible for inclusions common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutation, and transgenic mice expressing human SOD1 gene mutation. Neuropathology 2001; 21: 67–81
Oosthuyse B, Moons L, Storkebaum E, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 2001; 28: 131–8
Olney JW. Excitatory amino acids and neuropsychiatric disorders. Biol Psychiatry 1989; 26: 505–25
Choi DW. Excitotoxic cell death. J Neurol 1992; 23: 1261–76
Regan RF, Panter S, Witz A, et al. Ultrasructure of excitotoxic neuronal death in murine cortical culture. Brain Res 1995; 705: 188–9
Atlante A, Calissano P, Bobba A, et al. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett 2001; 497: 1–5
Leigh PN. Excitotoxicity in ALS. Neurology 1996; 47 Suppl. 4: S221–7
Ince PG, Eggett CJ, Shaw PJ. The role of excitotoxicity in neurological disease. Rev Contemp Pharmacol 1997; 8: 195–212
Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther 1999;81: 163–221
Jackson M, Rothsrein JD. Excitotoxicity in amyotrophic lateral sclerosis. In: Brown Jr RH, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz, 2000: 263–77
Van Den Bosch L, Vandenberghe W, Klaassen H, et al. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J Neurol Sci 2000; 180: 29–34
Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65: 1–105
Rothstein JD, Van Kammen M, Levey AI, et al. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 1995; 38: 73–84
Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 1996; 39: 676–9
Lin CL, Bristol LA, Jin L, et al. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter in amyotrophic lateral sclerosis. Neuron 1998; 20: 589–602
Meyer T, Munch C, Knappenberger B, et al. Alternative splicing of the glutamate transporter EAAT2 (GLT-1). Neurosci Lett 1998; 241: 68–70
Nagai M, Abe K, Okamoto K, et al. Identification of alternative splicing forms of GLT-1 mRNA in the spinal cord of amyotrophic lateral sclerosis patients. Neurosci Lett 1998; 244: 165–8
Jackson M, Steers G, Leigh PN, et al. Polymorphisms in the glutamate transporter gene EAAT2 in European ALS patients. J Neurol 1999; 246: 1140–4
Meyer T, Fromm A, Munch C, et al. The RNA of the glutamate transporter EAAT2 is variably spliced in amyotrophic lateral sclerosis and normal individuals. J Neurol Sci 1999; 170: 45–50
Honig LS, Chambliss DD, Bigio EH, et al. Glutamate transporter EAAT2 splice variants occur not only in ALS, but also in AD and controls. Neurology 2000; 55: 1082–8
Flowers JM, Powell JF, Leigh PN, et al. Intron 7 retention and exon 9 skipping EAAT2 mRNA variants are not associated with amyotrophic lateral sclerosis. Ann Neurol 2001; 49: 643–9
Aoki M, Lin CL, Rothstein JD, et al. Mutations in the glutamate transporter EAAT2 gene do not cause abnormal EAAT2 transcripts in amyotrophic lateral sclerosis. Ann Neurol 1998; 43: 645–53
Alexander GM, Deitch JS, Seeburger JL, et al. Elevated cortical extracellular fluid glutamate in transgenic mice expressing human mutant (G93A) Cu/Zn Superoxide dismutase. J Neurochem 2000; 74: 1666–73
Andreassen OA, Jenkins BG, Dedeoglu A, et al. Increases in cortical glutamate concentrations in transgenic amyotrophic lateral sclerosis mice are attenuated by creatine supplementation. J Neurochem 2001; 77: 383–90
Guo Z, Kindy MS, Kruman I, et al. ALS-linked Cu/Zn-SOD mutation impairs cerebral synaptic glucose and glutamate transport and exacerbates ischemic brain injury. J Cereb Blood Flow Metab 2000; 20: 463–8
Canton T, Pratt J, Stutzmann JM, et al. Glutamate uptake is decreased tardively in the spinal cord of FALS mice. Neuroreport 1998; 9: 775–8
Gurney ME, Cutting FB, Zhai P, et al. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol 1996; 39: 147–57
Gurney ME, Fleck TJ, Himes CS, et al. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology 1998; 50: 62–6
Canton T, Bohme GA, Boireau A, et al. RPR 119990, a novel alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid antagonist: synthesis, pharmacological properties, and activity in an animal model of amyotrophic lateral sclerosis. J Pharmacol Exp Ther 2001; 299: 314–22
Howland DS, Liu J, She Y, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 2002; 99: 1604–9
Benzie IF. Lipid peroxidation: a review of causes, consequences, measurement and dietary influences. Int J Food Sci Nutr 1996; 47: 233–61
Onorato JM, Thorpe SR, Baynes JW. Immunohistochemial and ELIS A assays for biomarkers of oxidative stress in aging and disease. Ann N Y Acad Sci 1998; 20: 277–90
Keller JN, Mattson MP. Roles of lipid peroxidation in modulation of cellular signalling pathways, cell dysfunction and death in the nervous system. Rev Neurosci 1998; 9: 105–60
Cookson MR, Shaw PJ. Oxidative stress and motor neurone disease. Brain Pathol 1999; 9: 135–86
Torreilles F, Salman-Tabcheh S, Guerin M, et al. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev 1999; 30: 153–6
Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev 1998; 78: 547–81
Floyd RA. Antioxidants, oxidative stress and degenerative neurological disorders. Proc Soc Exp Biol Med 1999; 222: 236–45
Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl 2000; 59: 133–54
Butterfield DA, Howard BJ, LaFontaine MA. Brain oxidative stress in animal models of accelerated aging and the age-related neurodegenerative disorders, Alzheimer's disease and Huntington's disease. Curr Med Chem 2001; 8: 815–28
Schapira AH. Causes of neuronal death in Parkinson's disease. Adv Neurol 2001; 86: 155–62
Andersen PM. Genetics of sporadic ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2 Suppl. 1: S37–42
Wiedau-Pazos M, Goto J, Rabizadeh S, et al. Altered reactivity of Superoxide dismutase in familial amyotrophic lateral sclerosis. Science 1996; 271: 515–8
Yim MB, Kang J-H, Yim H-S. A gain-of-function for an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci US A 1996; 93: 5709–14
Bogdanov MB, Ramos LE, Xu Z, et al. Elevated “hydroxyl radical” generation in vivo in an animal model of amyotrophic lateral sclerosis. J Neurochem 1998; 71: 1321–4
Crow JP, Sampson JB, Zhuang YX, et al. Decreased zinc affinity of amyotrophic lateral sclerosis-associated Superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem 1997; 69: 1936–44
Ogawa Y, Kosaka H, Nakanishi T, et al. Stability of mutant Superoxide dismutase-1 associated with familial amyotrophic lateral sclerosis determines the manner of copper release and induction of thioredoxin in erythrocytes. Biochem Biophys Res Commun 1997; 241: 251–7
Yim HS, Kang JH, Chock PB, et al. A familial amyotrophic lateral sclerosis-associated A4V Cu, Zn-superoxide dismutase mutant has a lower Km for hydrogen peroxide. J Biol Chem 1997; 272: 8861–3
Kim SM, Eum WS, Kwon OB, et al. The free radical-generating function of a familial amyotrophic lateral sclerosis-associated D90A Cu,Zn-superoxide dismutase mutant. Biochem Mol Biol Int 1998; 46: 1191–200
Estevez A, Crow JP, Sampson JB, et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient Superoxide dismutase. Science 1999; 286: 2498–500
Eum WS, Kang JH. Release of copper ions from the familial amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutants. Mol Cells 1999; 9: 110–4
Gabbianelli R, Ferri A, Rotilio G, et al. Aberrant copper chemistry as a major mediator of oxidative stress in a human cellular model of amyotrophic lateral sclerosis. J Neurochem 1999; 73: 1175–80
Goto JJ, Zhu H, Snachez RJ, et al. Loss of in vitro metal ion binding specificity in mutant copper-zinc Superoxide dismutase associated with familial amyotrophic lateral sclerosis. J Biol Chem 2000; 275: 1007–14
Kang JH, Eum WS. Enhanced oxidative damage by the familial amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutants. Biochim Biophys Acta 2000; 1524: 162–70
Lyons TJ, Nersissian A, Huang H, et al. The metal binding properties of the zinc site of yeast copper-zinc Superoxide dismutase: implications for amyotrophic lateral sclerosis. J Biol Inorg Chem 2000; 5: 189–203
Briujn LI, Houseweart MK, Kato S, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independant from wild-type SOD1. Science 1998; 281: 1851–4
Johnston JA, Dalton MJ, Gurney ME, et al. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2000; 97: 12571–6
Okado-Matsumoto A, Myint T, Fujii J, et al. Gain in function of mutant Cu, Zn-superoxide dismutases as a causative factor in familial amyotrophic lateral sclerosis: less reactive oxidant formation but high spontaneous aggregation and precipitation. Free Radic Res 2000; 33: 65–73
Kato S, Sumi-Akamaru H, Fujimura H, et al. Copper chaperone for Superoxide dismutase co-aggregates with Superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutation. Acta Neuropathol (Berl) 2001; 102: 233–8
Oeda T, Shimohama S, Kitagawa N, et al. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum Mol Genet 2001; 10: 2013–23
Julien JP. Amyotrophic lateral sclerosis, unfolding the toxicity of the misfolded. Cell 2001; 104: 581–91
Bowling AC, Schulz JB, Brown RHJ, et al. Superoxide dismutase activity, oxidative damage and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem 1993; 61: 2322–5
Shaw PJ, Ince PG, Falkous G, et al. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann Neurol 1995; 38: 691–5
Abe K, Pan LH, Watanabe M, et al. Upregulation of protein-tyrosine nitration in the anterior horn of cells of amyotrophic lateral sclerosis. Neurol Res 1997; 19: 124–8
Beal MF, Ferrante RJ, Browne SE, et al. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol 1997; 42: 646–54
Ferrante RJ, Browne SE, Shinobu LA, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem 1997; 69: 2064–74
Oteiza PI, Uchitel OD, Carrasquedo F, et al. Evaluation of antioxidants, protein and lipid oxidation in blood from sporadic amyotrophic lateral sclerosis patients. Neurochem Res 1997; 22: 535–9
Smith G, Henry YK, Mattson MP, et al. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol 1998; 44: 696–9
Borthwick GM, Johnson MA, Ince PG, et al. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol 1999; 46: 787–90
Toghi H, Abe T, Yamazaki K, et al. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with amyotrophic lateral sclerosis. Ann Neurol 1999; 46: 129–31
Bogdanov M, Brown RH, Matson W, et al. Increased oxidative damage to DNA in ALS patients. Free Radie Biol Med 2000; 29: 652–8
Aoyama K, Matsubara K, Fujikawa Y, et al. Nitration of manganese Superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann Neurol 2000; 47: 524–7
Bonnefont-Rousselot D, Lacomblez L, Jaudon M, et al. Blood oxidative stress in amyotrophic lateral sclerosis. J Neurol Sci 2000; 178: 57–62
Sasaki S, Shibata N, Komori T, et al. iNOS and nitrotyrosine immunoreactivity in amyotrophic lateral sclerosis. Neurosci Lett 2000; 291: 44–8
Shibata N, Nagai R, Uchida K, et al. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res 2001; 917: 97–104
Ferrante RJ, Shinobu LA, Schulz JB, et al. Increased 3-nitrotyrosine and oxidative damage in mice with a copper/zinc Superoxide dismutase mutation. Ann Neurol 1997; 42: 326–34
Oostveen JA, Gurney ME, Hall ED. Immunocytochemical evidence of spinal cord peroxidation, peroxynitrite formation and astrocyte and microglial activation in the transgenic model of familial amyotrophic lateral sclerosis [abstract]. Soc Neurosci Abs 1997; 23: 13
Andrus PK, Andersen PM, Nilsson P, et al. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 1998; 71: 2041–8
Hall ED, Andrus PK, Oostveen JA, et al. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J Neurosci Res 1998; 53: 66–77
Liu D, Wen J, Liu J, et al. The roles of free radicles in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA and membrane phospholipids. FASEB J 1999; 13: 2318–28
Cha CI, Chung YH, Shin C, et al. Immunocytochemical study on the distribution of nitrotyrosine in the brain of the transgenic mice expressing a human Cu/Zn SOD mutation. Brain Res 2000; 853: 156–61
Warita H, Hayashi T, Murakami T, et al. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res Mol Brain Res 2001; 89: 147–52
Dugan LL, Turetsky DM, Du C, et al. Carboxyfullerenes as neuroprotective agents. Proc Natl Acad Sci U S A 1997; 94: 9434–9
Poduslo JF, Whelan SL, Curran GL, et al. Therapeutic benefit of polyamine-modified catalase as a scavenger of hydrogen peroxide and nitric oxide in familial amyotrophic lateral sclerosis transgenics. Ann Neurol 2000; 48: 943–7
Reinholz MM, Merkle CM, Poduslo JF. Therapeutic benefits of putrescine-modified catalase in a transgenic mouse model of familial amyotrophic lateral sclerosis. Exp Neurol 1999; 159: 204–16
Jung C, Rong Y, Doctrow S, et al. Synthetic Superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci Lett 2001; 304: 157–60
Jaarsma D, Guchelaar HJ, Haasdijk E, et al. The antioxidant N-acetylcysteine does not delay disease onset and death in a transgenic mouse model of amyotrophic lateral sclerosis [abstract]. Ann Neurol 1998; 44: 293
Andreassen OA, Dedeoglu A, Klivenyi P, et al. N-acetyl-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport 2000; 11: 2491–3
Hagen TM, Ingersoll RT, Lykkesfeldt J, et al. (R)-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J 1999; 13: 411–8
Andreassen OA, Dedeoglu A, Friedlich A, et al. Effects of an inhibitor of poly(ADP-ribose) polymerase, desmethylselegiline, trientine, and lipoic acid in transgenic ALS mice. Exp Neurol 2001; 168: 419–24
Barneoud P, Curet O. Beneficial effects of lysine acetylsalicylate, a soluble salt of aspirin, on motor performance in a transgenic model of amyotrophic lateral sclerosis. Exp Neurol 1999; 155: 243–51
Jiang F, DeSilva S, Turnbull J. Beneficial effect of ginseng root in SOD-1 (G93A) transgenic mice. J Neurol Sci 2000; 180: 52–4
Facchinetti F, Sasaki M, Cutting FB, et al. Lack of involvement of neuronal nitric oxide synthase in the pathogenesis of a transgenic mouse model of familial amyotrophic lateral sclerosis. Neuroscience 1999; 90: 1483–92
Upton-Rice MN, Cudkowicz ME, Mathew RK, et al. Administration of nitric oxide synthase inhibitors does not alter disease course of amyotrophic lateral sclerosis SOD 1 mutant transgenic mice. Ann Neurol 1999; 45: 413–4
Aimer G, Vukosavic S, Romero N, et al. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 1999; 72: 2415–25
Ferrante RJ, Klein AM, Dedeoglu A, et al. Therapeutic efficacy of EGb761 (Gingko biloba extract) in a transgenic mouse model of amyotrophic lateral sclerosis. J Mol Neurosci 2001; 17: 89–96
Hottinger AF, Fine EG, Gurney ME, et al. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur J Neurosci 1997; 9: 1548–51
Nagano S, Ogawa Y, Yanagihara T, et al. Benefit of a combined treatment with trientine and ascorbate in familial amyotrophic lateral sclerosis model mice. Neurosci Lett 1999; 265: 159–62
Nagano S, Satoh M, Sumi H, et al. Reduction of metal-lothioneins promotes the disease expression of familial amyotrophic lateral sclerosis mice in a dose-dependent manner. Eur J Neurosci 2001; 13: 1363–70
Subramaniam JR, Lyons WE, Liu J, et al. Mutant SOD 1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci 2002; 5: 301–7
Gurney ME. Transgenic models of familial amyotrophic lateral sclerosis. J Neurol 1997; 244 Suppl. 2: S15–20
Trotti D, Rolfs A, Danbolt NC, et al. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci 1999; 2: 427–33
Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochem Biophys Acta 1998; 1366: 211–23
Browne SE, Bowling AC, Baik MJ, et al. Metabolic dysfunction in familial, but not sporadic amypotrophic lateral sclerosis. J Neurochem 1998; 71: 281–7
Swerdlow RH, Parks JK, Cassarino DS, et al. Mitochondria in sporadic amyotrophic lateral sclerosis. Exp Neurol 1998; 153: 135–42
Wiedemann FR, Winkler K, Kuznetsov AV, et al. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci 1998; 156: 65–72
Dhaliwal GK, Grewal RP. Mitochondrial DNA deletion mutation levels are elevated in ALS brains. Neuroreport 2000; 11: 2507–9
Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci 2000; 23: 298–304
Swerdlow RH, Parks JK, Pattee G, et al. Role of mitochondria in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 185–90
Wong PC, Pardo CA, Borchelt DR, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995; 14: 1105–16
Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice carrying a mutant SOD1. J Neurosci 1998; 18: 3241–50
Andreassen OA, Ferrante RJ, Klivenyi P, et al. Transgenic ALS mice show increased vulnerability to the mitochondrial toxins MPTP and 3-nitropropionic acid. Exp Neurol 2001; 168: 356–63
Andreassen OA, Ferrante RJ, Klivenyi P, et al. Partial deficiency of manganese Superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann Neurol 2000; 47: 447–55
Matthews RT, Yang L, Browne S, et al. Coenzyme Q administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A 1998; 95: 8892–7
Beal MF. Coenzyme Q10 administration and its potential for treatment of neurodegenerative diseases. Biofactors 1999; 9: 261–6
Kliveny P, Ferrante RJ, Matthews RT, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nature Med 1999; 5: 347–50
Tarnopolsky MA, Beal MF. Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann Neurol 2001; 49: 561–73
Sathasivam S, Ince PG, Shaw PJ. Apoptosis in amyotrophic lateral sclerosis: a review of the evidence. Neuropathol Appl Neurobiol 2001; 27: 257–74
Bratton SB, Cohen GM. Apoptotic death sensor: an organelle's alter ego? Trends Pharmacol Sci 2001; 22: 306–15
Warita H, Manabe Y, Murakami T, et al. Early decrease of survival signal-related proteins in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Apoptosis 2001; 6: 345–52
Guegan C, Vila M, Rosoklija G, et al. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 2001; 21: 6569–76
de la Monte SM, Sohn YK, Ganju N, et al. P53- and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest 1998; 78: 401–11
Martin LJ. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis 2000; 7: 613–22
Mu X, He J, Anderson DW, et al. Altered expression of bcl-2 and bax mRNA in amyotrophic lateral sclerosis spinal cord motor neurons. Ann Neurol 1996; 40: 379–86
Tews DS, Goebel HH, Meinck HM. DNA-fragmentation and apoptosis-related proteins of muscle cells in motor neuron disorders. Acta Neurol Scand 1997; 96: 380–6
Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 1999; 58: 459–71
Ekegren T, Grundstrom E, Lindholm D, et al. Upregulation of Bax protein and increased DNA degradation in ALS spinal cord motor neurons. Acta Neurol Scand 1999; 100: 317–21
Schoser BG, Wehling S, Blottner D. Cell death and apoptosis-related proteins in muscle biopsies of sporadic amyotrophic lateral sclerosis and polyneuropathy. Muscle Nerve 2001; 24: 1083–9
Shinoe T, Wanaka A, Nikaido T, et al. Upregulation of the pro-apoptotic BH3-only peptide harakiri in spinal neurons of amyotrophic lateral sclerosis patients. Neurosci Lett 2001; 313: 153–7
Pedersen WA, Luo H, Kruman I, et al. The prostate apoptosis response-4 protein participates in motor neuron degeneration in amyotrophic lateral sclerosis. FASEB J 2000; 14: 913–24
Yuan J, Yanker BA. Apoptosis in the nervous system. Nature 2000; 407: 802–9
Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature 2000; 407: 810–6
Li M, Ona VO, Guegan C, et al. Functional role of capsase-1 and capsase-3 in an ALS transgenic mouse model. Science 2000; 288: 335–9
Ilzecka J, Stelmasiak Z, Dobosz B. Interleukin-lbeta converting enzyme/Caspase-1 (ICE/Caspase-1) and soluble APO-1/Fas/CD 95 receptor in amyotrophic lateral sclerosis patients. Acta Neurol Scand 2001; 103: 255–8
Migheli A, Atzori C, Piva R, et al. Lack of apoptosis in mice with ALS. Nat Med 1999; 5: 966–7
Spooren WP, Hengerer B. DNA laddering and caspase 3-like activity in the spinal cord of a mouse model of familial amyotrophic lateral sclerosis. Cell Mol Biol (Noisy-le-grand) 2000; 46: 63–9
Vukosavic S, Dubois-Dauphin M, Romero N, et al. Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 1999; 73: 2460–8
Kostic V, Jackson-Lewis V, de Bilbao F, et al. Bcl-2: prolonging life in a transgenic mouse model of amyotrophic lateral sclerosis. Science 1997; 277: 559–62
Azzouz M, Hottinger A, Paterna JC, et al. Increased motorneuron survival and improved neuromusclar function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum Mol Genet 2000; 9: 803–11
Pasinelli P, Houseweart MK, Brown RHJ, et al. Caspase-1 and-3 are sequentially activated in motor neuron death in Cu,Zn Superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2000; 97: 13901–6
Vukosavic S, Stefanis L, Jackson-Lewis V, et al. Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 2000; 20: 9119–25
Friedlander RM, Brown RH, Gagliardini V, et al. Inhibition of ICE slows ALS in mice [letter]. Nature 1997; 388: 31
Trieu VN, Lui R, Liu X-P, et al. A specific inhibitor of Janus kinase-3 increases survival in a transgenic mouse model of amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2000; 267: 22–5
Goodman PA, Niehoff LB, Uckun FM. Role of tyrosine kinases in induction of the c-jun proto-oncogene in irradiated B-lineage lymphoid cells. J Biol Chem 1998; 273: 27028–38
Keep M, Elmer E, Fong KS, et al. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res 2001; 894: 327–31
Chen M, Ona VO, Li M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000; 6: 797–801
Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002; 417: 74–8
Gonzalez de Aguilar JL, Gordon JW, Rene F, et al. Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: evidence for the implication of the p53 signaling pathway. Neurobiol Dis 2000; 7: 406–15
Kuntz C, Kinoshita Y, Beal MF, et al. Absence of p53: no effect in a transgenic mouse model of familial amyotrophic lateral sclerosis. Exp Neurol 2000; 165: 184–90
Prudlo J, Koenig J, Graser J, et al. Motor neuron cell death in a mouse model of FALS is not mediated by the p53 cell survival regulator. Brain Res 2000; 879: 183–7
Aimer G, Guegan C, Teismann P, et al. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol 2001; 49: 176–85
Frank KM, Coccia C, Drachman DB, et al. COX-2 inhibition prolongs survival in atransgenic mouse model of ALS [abstract]. Soc Neurosci Abstracts 2001; 2001: 296
Mohajeri MH, Figlewicz DA, Bohn MC. Intramuscular grafts of myoblasts genetically modified to secrete glial cell line-derived neurotrophic factor prevent motoneuron loss and disease progression in a mouse model of familial amyotrophic lateral sclerosis. Hum Gene Ther 1999; 10: 1853–66
Bordet T, Lesbordes JC, Rouhani S, et al. Protective effects of cardiotrophin-1 adenoviral gene transfer on neuromuscular degeneration in transgenic ALS mice. Hum Mol Genet 2001; 10: 1925–33
Upton-Rice MN, Cudkowicz ME, Warren L, et al. Basic fibro-blast growth factor does not prolong survival in a transgenic model of familial amyotrophic lateral sclerosis [abstract]. Ann Neurol 1999; 46: 934
Leigh PN, Swash M. Cytoskeletal pathology in motor neuron diseases. Adv Neurol 1991; 56: 115–24
Figlewicz DA, Krizus A, Martinoli MG, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet 1994; 3: 1757–61
Al-Chalabi A, Andersen PM, Nilsson P, et al. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet 1999; 8: 157–64
Julien JP, Beaulieu JM. Cytoskeletal abnormalities in amyotrophic lateral sclerosis: beneficial or detrimental effects? J Neurol Sci 2000; 180: 7–14
Lee MK, Cleveland DW. Neuronal intermediate filaments. Annu Rev Neurosci 1996; 19: 187–217
Beaulieu JM, Nguyen MD, Julien JP. Late onset death of motor neurons in mice overexpressing wild-type peripherin. J Cell Biol 1999; 147: 531–44
Al-Chalabi A, Leigh PN. Recent advances in amyotrophic lateral sclerosis. Curr Opin Neurol 2000; 13: 397–405
Rowland LP. Six important themes in amyotrophic lateral sclerosis (ALS) research, 1999. J Neurol Sci 2000; 180: 2–6
Hand CK, Rouleau GA. Familial amyotrophic lateral sclerosis. Muscle Nerve 2002; 25: 135–59
Morrison KE. Therapies in amyotrophic lateral sclerosis-beyond riluzole. Curr Opin Pharmacol 2002; 2: 302–9
Chen R, Ende N. The potential for the use of mononuclear cells from human umbilical cord blood in the treatment of amyotrophic lateral sclerosis in SOD 1 mice. J Med 2000; 31: 21–30
Ende N, Weinstein F, Chen R, et al. Human umbilical cord blood effect on sod mice (amyotrophic lateral sclerosis). Life Sci 2000; 67: 53–9
Anneser JM, Gmerek A, Gerkrath J, et al. Immunosuppressant FK506 does not exert beneficial effects in symptomatic G93A Superoxide dismutase-1 transgenic mice. Neuroreport 2001; 12: 2663–5
Miller RG, Moore D, Young A. Placebo-controlled trial of gabapentin in patients with amyotrophic lateral sclerosis: WALS Study Group. Neurology 1996; 47: 1383–8
Miller RG, Moore 2nd DH, Gelinas DF, et al. Phase III randomized trial of gabapentin in patients with amyotrophic lateral sclerosis. Neurology 2001; 56: 843–8
Blin O, Pouget J, Aubrespy G, et al. A double-blind placebo-controlled trial of L-threonine in amyotrophic lateral sclerosis. J Neurol 1992; 239: 79–81
Testa D, Caraceni T, Fetoni V, et al. Chronic treatment with L-threonine in amyotrophic lateral sclerosis: a pilot study. Clin Neurol Neurosurg 1992; 94: 7–9
Group TIAS. Branched-chain amino acids and amyotrophic lateral sclerosis: a treatment failure? Neurology 1993; 43: 2466–70
Tandan R, Bromberg MB, Forshew D, et al. A controlled trial of amino acid therapy in amyotrophic lateral sclerosis: I. clinical, functional, and maximum isometric torque data. Neurology 1996; 47: 1220–6
Eisen A, Stewart H, Schulzer M, et al. Anti-glutamate therapy in amyotrophic lateral sclerosis: a trial using lamotrigine. Can J Neurol Sci 1993; 20: 297–301
Askmark H, Aquilonius SM, Gillberg PG, et al. A pilot trial of dextromethorphan in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 1993; 56: 197–200
Gredal O, Werdelin L, Bak S, et al. A clinical trial of dextromethorphan in amyotrophic lateral sclerosis. Acta Neurol Scand 1997; 96: 8–13
Blin O, Azulay JP, Desnuelle C, et al. A controlled one-year trial of dextromethorphan in amyotrophic lateral sclerosis. Clin Neuropharmacol 1996; 19: 189–92
Miller RG, Shepherd R, Dao H, et al. Controlled trial of nimodipine in amyotrophic lateral sclerosis. Neuromuscul Disord 1996; 6: 101–4
Miller RG, Smith SA, Murphy JR, et al. A clinical trial of verapamil in amyotrophic lateral sclerosis. Muscle Nerve 1996; 19: 511–5
Louwerse ES, Weverling GJ, Bossuyt PMM. Randomised, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol 1995; 52: 559–64
Chio A, Cucatto A, Terreni AA, et al. Reduced glutathione in amyotrophic lateral sclerosis: an open, crossover, randomized trial. Ital J Neurol Sci 1998; 19: 363–6
Mazzini L, Testa D, Balzarini C, et al. An open-randomized clinical trial of selegiline in amyotrophic lateral sclerosis. J Neurol 1994; 241: 223–7
Jossan SS, Ekblom J, Gudjonsson O, et al. Double blind cross over trial with deprenyl in amyotrophic lateral sclerosis. J Neural Transm Suppl 1994; 41: 237–41
Lange DJ, Murphy PL, Diamond B, et al. Selegiline is ineffective in a collaborative double-blind, placebo-controlled trial for treatment of amyotrophic lateral sclerosis. Arch Neurol 1998; 55: 93–6
Desnuelle C, Dib M, Garrel C, et al. A double-blind, placebocontrolled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis: ALS riluzole-tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2: 9–18
Bradley WG. A phase I/II study of recombinant brain-derived neutrotrophic factor in patients with ALS [abstract]. Ann Neurol 1995; 38: 971
Group TBS. A controlled trial ofrecombinant methionyl human BDNF in ALS: The BDNF Study Group (Phase III). Neurology 1999; 52: 1427–33
ALS CNTF Treatment Study Group. A double-blind placebocontrolled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. ALS CNTF Treatment Study Group. Neurology 1996; 46: 1244–9
Miller RG, Petajan JH, Bryan WW, et al. A placebo-controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis. rhCNTF ALS Study Group. Ann Neurol 1996; 39: 256–60
Lai EC, Felice KJ, Festoff BW, et al. Effect of recombinant human insulin-like growth factor-I on progression of ALS: a placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology 1997; 49: 1621–30
Borasio GD, Robberecht W, Leigh PN, et al. A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis: European ALS/IGF-I Study Group. Neurology 1998; 51: 583–6
Smith RA, Melmed S, Sherman B, et al. Recombinant growth hormone treatment of amyotrophic lateral sclerosis. Muscle Nerve 1993; 16: 624–33
Munsat TL, Taft J, Jackson IM, et al. Intrathecal thyrotropin-releasing hormone does not alter the progressive course of ALS: experience with an intrathecal drug delivery system. Neurology 1992; 42: 1049–53
Beghi E, Chio A, Inghilleri M, et al. A randomized controlled trial of recombinant interferon beta-1a in ALS: Italian Amyotrophic Lateral Sclerosis Study Group. Neurology 2000; 54: 469–74
Gourie-Devi M, Nalini A, Subbakrishna DK. Temporary amelioration of symptoms with intravenous cyclophosphamide in amyotrophic lateral sclerosis. J Neurol Sci 1997; 150: 167–72
Smith SA, Miller RG, Murphy JR, et al. Treatment of ALS with high dose pulse cyclophosphamide. J Neurol Sci 1994; 124 Suppl.: 84–7
Drachman DB, Chaudhry V, Cornblath D, et al. Trial of immunosuppression in amyotrophic lateral sclerosis using total lymphoid irradiation. Ann Neurol 1994; 35: 142–50
Meucci N, Nobile-Orazio E, Scarlato G. Intravenous immunoglobulin therapy in amyotrophic lateral sclerosis. J Neurol 1996; 243: 117–20
Dalakas MC, Stein DP, Otero C, et al. Effect of high-dose intravenous immunoglobulin on amyotrophic lateral sclerosis and multifocal motor neuropathy. Arch Neurol 1994;51: 861–4
Aisen ML, Sevilla D, Edelstein L, et al. A double-blind placebo-controlled study of 3,4-diaminopyridine in amytrophic lateral sclerosis patients on a rehabilitation unit. J Neurol Sci 1996; 138: 93–6
Kaji R, Kodama M, Imamura A, et al. Effect of ultrahigh-dose methylcobalamin on compound muscle action potentials in amyotrophic lateral sclerosis: a double-blind controlled study. Muscle Nerve 1998; 21: 1775–8
Cudkowicz ME, Warren L, Francis JW, et al. Intrathecal administration of recombinant human Superoxide dismutase 1 in amyotrophic lateral sclerosis: a preliminary safety and pharmacokinetic study. Neurology 1997; 49: 213–22
Koliatsos VE, Clatterbuck RE, Winslow JW, et al. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron 1993; 10: 359–67
Kishino A, Ishige Y, Tatsuno T, et al. BDNF prevents and reverses adult rat motor neuron degeneration and induces axonal outgrowth. Exp Neurol 1997; 144: 273–86
Mitsumoto H, Ikeda K, Klinkosz B, et al. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science 1994; 265: 1107–10
Ikeda K, Klinkosz B, Greene T, et al. Effects of brain-derived neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann Neurol 1995; 37: 505–11
Tsuzaka K, Ishiyama T, Pioro EP, et al. Role of brain-derived neurotrophic factor in wobbler mouse motor neuron disease. Muscle Nerve 2001; 24: 474–80
ALS association website: drug clinical news. Arugen-Regeneron partners discontinuing all cinical development of BDNF. Available from URL: http://www.alsa.org/news/news012801.cfm [Accessed 2002 Nov 20]
Sagot Y, Tan SA, Baetge E, Schmalbruch H, Kato AC, Aebischer Polymer encapsulated cell lines genetically engineered to release ciliary neurotrophic factor can slow down progressive motor neuronopathy in the mouse. Eur J Neurosci 1995; 7: 1313–1322
Hantai D, Akaaboune M, Lagord C, et al. Beneficial effects of insulin-like growth factor-I on wobbler mouse motoneuron disease. J Neurol Sci 1995; 129 Suppl.: 122–6
Armon C, Graves MC, Moses D, et al. Linear estimates of disease progression predict survival in patients with amyotrophic lateral sclerosis. Muscle Nerve 2000; 23: 874–82
Duong FH, Warter JM, Poindron P, et al. Effect of the nonpeptide neurotrophic compound SR 57746A on the phenotypic survival of purified mouse motoneurons. Br J Pharmacol 1999; 128: 1385–92
Iwasaki Y, Shiojima T, Kinoshita M, et al. SR57746A: a survival factor for motor neurons in vivo. JNeurolSci 1998; 160 Suppl. l: S92–6
Duong F, Fournier J, Keane PE, et al. The effect of the nonpeptide neurotrophic compound SR 57746A on the progression of the disease state of the pmn mouse. Br J Pharmacol 1998; 124: 811–7
Sanofi-Synthelabous US affiliate site: press releases. First half 2000 results. Available from URL: www.sanofi-synthelabous.com/news/20000906.htm [Accessed 2002 Nov 20]
Rosenfeld WE. Topiramate: a review of preclinical, pharmacokinetic, and clinical data. Clin Ther 1997; 19: 1294–308
Jackson PF, Slusher BS. Design of naaladase inhibitors: anovel neuroprotective strategy. Curr Med Chem 2001; 8: 949–57
Mazzini L, Balzarini C, Colombo R, et al. Effects of creatine supplementation on exercise performance and muscular strength in amyotrophic lateral sclerosis: preliminary results. J Neurol Sci 2001; 191: 139–44
Bronowska A, Les A, Chilmonczyk Z, et al. Molecular dynamics of buspirone analogues interacting with the 5-HT1A and 5-HT2A serotonin receptors. Bioorg Med Chem 2001; 9: 881–95
Neotrofin™. Available from URL: http://www.alsa.org/research/drugdev9.cfm [Accessed 2002 Nov 20]
Moulignier A, Moulonguet A, Pialoux G, et al. Reversible ALS-like disorder in HIV infection. Neurology 2001; 57: 995–1001
MacGowan DJ, Scelsa SN, Waldron M. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology 2001; 57: 1094–7
Louvel E, Hugon J, Doble A. Therapeutic advances in amyotrophic lateral sclerosis. Trends Pharmacol Sci 1997;18: 196–203
Turner MR, Parton MJ, Leigh PN. Clinical trials in ALS: an overview. Semin Neurol 2001; 21: 167–75
Munsat TL. Issues in amyotrophic lateral sclerosis clinical trial design. Adv Neurol 1995; 68: 209–18
Meininger V, Salachas F. Review of clinical trials. In: Brown Jr RH, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz, 2000: 389–402
Meininger V. Clinical trials: the past, a lesson for the future. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2 Suppl. 1: S15–8
Mitsumoto H. Clinical trials: present and future. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2 Suppl. 1: S10–4
Miller RG, Munsat TL, Swash M, et al. Consensus guidelines for the design and implementation of clinical trials in ALS: World Federation of Neurology committee on Research. J Neurol Sci 1999; 169: 2–12
World Federation of Neurology. Consensus guidelines for clinical trials in ALS. Available from URL: http://www.wfnals.org/Articles/airliel998guidelines.htm [Accessed 2002 Nov 20]
Gelinas D. Conceptual approach to diagnostic delay in ALS: a United States Perspective. Neurolgy 1999; 53 Suppl. 5: S17–9
Dengler R. Current treatment pathways in ALS: a European perspective. Neurology 1999; 53 Suppl. 5: S4–S10
Belsh JM. Diagnostic challenges in ALS. Neurology 1999; 53 Suppl. 5: S26–30
Brooks BR. What are the implications of early diagnosis?: maintaining optimal health as long as possible. Neurology 1999; 53 Suppl. 5: S43–5
Ludolph AC, Riepe MW. Do the benefits of currently available treatments justify early diagnosis and treatment of amyotrophic lateral sclerosis?: arguments against. Neurology 1999; 53: S46–9
Cashman NR. Do the benefits of currently available treatments justify early diagnosis and treatment of amyotrophic lateral sclerosis?: arguments for. Neurology 1999; 53: S50–2
Brooks BR. Earlier is better: the benefits of early diagnosis. Neurology 1999; 53 Suppl. 5: S53–4
Morrison BM, Janssen WG, Gordon JW, et al. Time course of neuropathology in the spinal cord of G86R Superoxide dismutase transgenic mice. J Comp Neurol 1999; 391: 64–77
Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD 1 mutants to motor neurons. Nat Neurosci 1999; 2: 50–6
Hansen S, Ballantyne JP. A quantitative electrophysiological study of motor neurone disease. J Neurol Neurosurg Psychiatry 1978; 41: 773–83
Sobue G, Sahashi K, Takahashi A, et al. Degenerating compartment and functioning compartment of motor neurons in ALS: possible process of motor neuron loss. Neurology 1983; 33: 654–7
Brooks BR, Sanjak M, Beiden D, Juhasz-Poscine K, Waclawik A. Natural history of amyotrophic lateral sclerosis-impairment, disability, handicap. In: Brown Jr RH, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz, 2000: 31–58
Shaw PJ, Williams R. Serum and cerebrospinal fluid biochemical markers of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1 Suppl. 2: S61–7
Dib M, Garrel C, Favier A, et al. Can Malondialdehyde be used as a biological marker of progression in neurodegenerative disease? J Neurol 2002; 249: 367–74
Karitzky J, Ludolph AC. Imaging and neurochemical markers for diagnosis and disease progression in ALS. J Neurol Sci 2001; 191: 35–41
Suhy J, Miller RG, Rule R, et al. Early detection and longitudinal changes in amyotrophic lateral sclerosis by (1)H MRSI. Neurology 2002; 58: 773–9
Bermejo P, Gomez-Serranillos P, Santos J, et al. Determination of malonaldehyde in Alzheimer's disease: a comparative study of high-performance liquid chromatography and thiobarbituric test. Gerontology 1997; 43: 218–22
Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol 1999; 56: 1449–52
Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem 2001; 8: 721–38
Andersen PM, Morita M, Brown RHJ. Genetics of amyotrophic lateral sclerosis: an overview. In: Brown Jr RH, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Martin Dunitz, 2000: 145–160
Vastag B. Stem cells step closer to the clinic: paralysis partially reversed in rats with ALS-like disease. JAMA 2001; 285: 1691–3
Acknowledgements
Dr Dib would like to thank Adam Doble and Ann Beaumont for their assistance in the preparation of this manuscript. Dr Dib is an employee of Aventis Pharma, the manufacturer of riluzole.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dib, M. Amyotrophic Lateral Sclerosis. Drugs 63, 289–310 (2003). https://doi.org/10.2165/00003495-200363030-00004
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003495-200363030-00004