Article Text

Abstract
Rupture of a saccular intracranial artery aneurysm (IA) causes subarachnoid hemorrhage, a significant cause of stroke and death. The current treatment options, endovascular coiling and clipping, are invasive and somewhat risky. Since only some IAs rupture, those IAs at risk for rupture should be identified. However, to improve the imaging of rupture-prone IAs and improve IA treatment, IA wall pathobiology requires more thorough knowledge. Chronic inflammation has become understood as an important phenomenon in IA wall pathobiology, featuring inflammatory cell infiltration as well as proliferative and fibrotic remodulatory responses. We review the literature on what is known about inflammation in the IA wall and also review the probable mechanisms of how inflammation would result in the degenerative changes that ultimately lead to IA wall rupture. We also discuss current options in imaging inflammation and how knowledge of inflammation in IA walls may improve IA treatment.
- Aneurysm
- inflammation
- intracranial aneurysm
Statistics from Altmetric.com
Introduction
The saccular intracranial artery aneurysm (IA) usually forms at cerebral artery bifurcations, but its etiology remains unknown. Recent studies on IAs have strengthened the concept that chronic inflammation predetermines IA wall degeneration toward a rupture-prone phenotype. In population-based studies in western countries, subarachnoid hemorrhage (SAH) caused by IA rupture comprises 1–7% of all strokes, as reviewed by Feigin et al.1 Approximately half of all patients with SAH die, and half of those surviving have problems in daily living.2 Current options to prevent the rupture or rerupture of an IA are clipping and endovascular therapy—both invasive and somewhat risky.3 According to a meta-analysis of studies published between 1955 and 1996, 2.3% of populations have been estimated to have IAs.4 Patients with aneurysmal SAH are hypertensive almost twice as often as non-aneurysmal SAH patients.5 Other known acquired risk factors for IAs are smoking and excessive alcohol consumption.6 Females are at higher risk for IA7 and IA rupture.4 Approximately 10% of IA patients have a family history of IA.8
Inflammation is a normal tissue's reaction to physical, chemical or biological stress or injury, manifesting in inflammatory cell accumulation and the immunological response. The classical clinical symptoms of an inflammatory reaction: rubor (redness), calor (heat), tumor (swelling) and dolor (pain), occur during microbial infections, where inflammation plays an important role as a defense mechanism against invading pathogens. However, in many acute and chronic inflammatory diseases, the inflammatory reaction is aseptic, involving no pathogen. Moreover, inflammation occurs in many diseases as a reaction to tissue injury caused by any mechanism, and as part of wound healing and scar formation.
Inflammation was first suggested to occur in IAs by Virchow in 1847.9 10 Further evidence comes from the 1930s when Maass11 12 described round-cell infiltration, most likely lymphocytic, especially in the IA neck. Later studies have shown the presence of inflammatory cell infiltrations and inflammatory mediators in the IA wall and finally their association with critical weakening of IA wall and wall rupture. A well-known but extremely rare cause of IA and its rupture is a local bacterial infection and an inflammatory response triggered by a local microbe, underlining the capacity of local inflammation to destroy tissue. However, this review focuses only on non-infectious, sporadic IAs.
We have previously studied the IA wall resected intraoperatively after IA clipping, focusing especially on the relation between IA histopathological features, inflammatory cell infiltration, complement activation, growth factor receptors, and IA rupture status and wall degeneration. We have also introduced an IA wall classification based on histological parameters that may aid in identification of those unruptured IAs susceptible to rupture.
We review what is known about IA pathobiology and the role of inflammation and also discuss the role of inflammation in probable biological processes leading to IA formation, growth and ultimately, rupture. We also hope to inspire clinicians and researchers by looking into future perspectives and prospects of potential anti-inflammatory treatment. To sustain readability, we describe most biological processes in a simplified manner.
Histopathology of IA wall degeneration
IA is formed at cerebral artery bifurcation
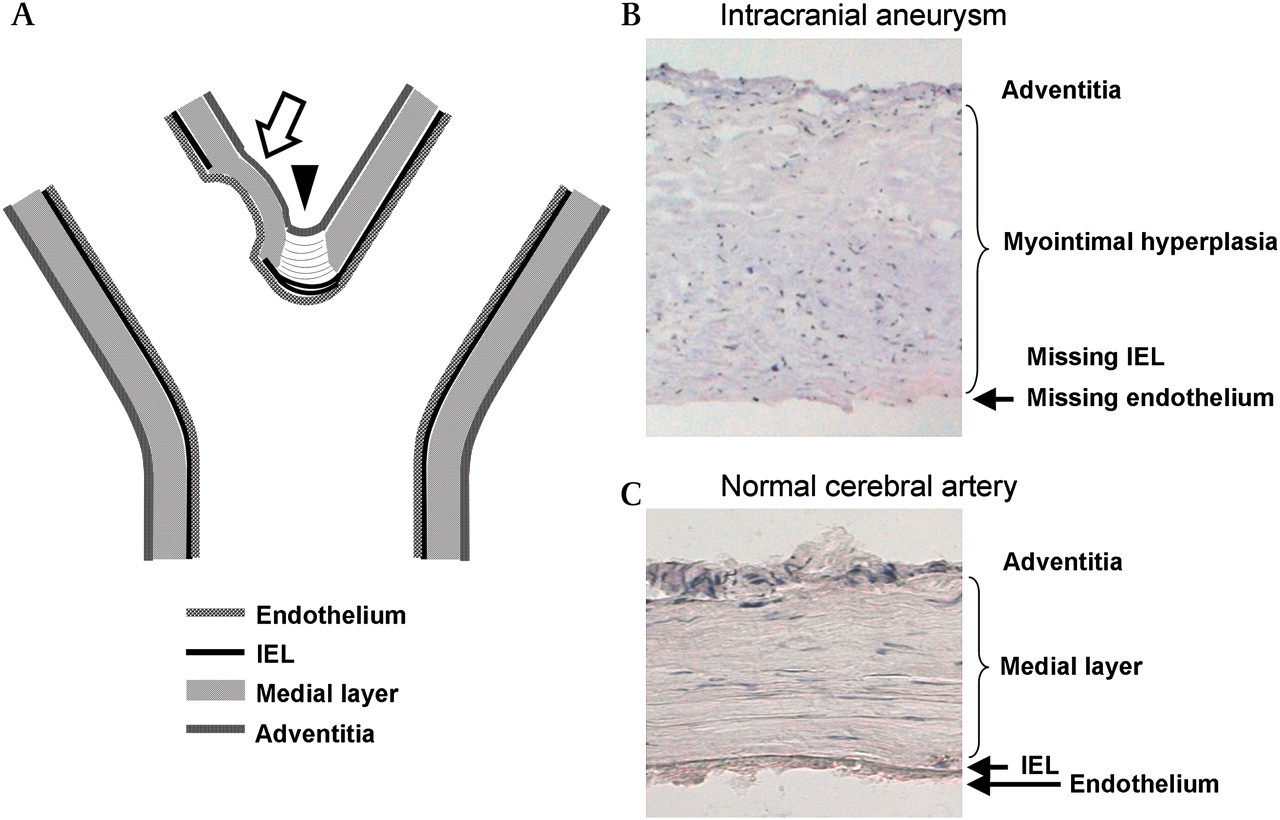
Tears in the internal elastic lamina (IEL) and associated vascular wall remodeling have been thought to precede IA formation, as the IA wall lacks the IEL, which normally provides structural strength for cerebral artery walls (figure 1). These changes have been detected especially at cerebral artery bifurcations of IA patients, but also occasionally at bifurcations in healthy individuals.12 In an experimental setting, tears in the IEL of a normal artery13 as well as advanced IAs14 can be induced by hemodynamics.
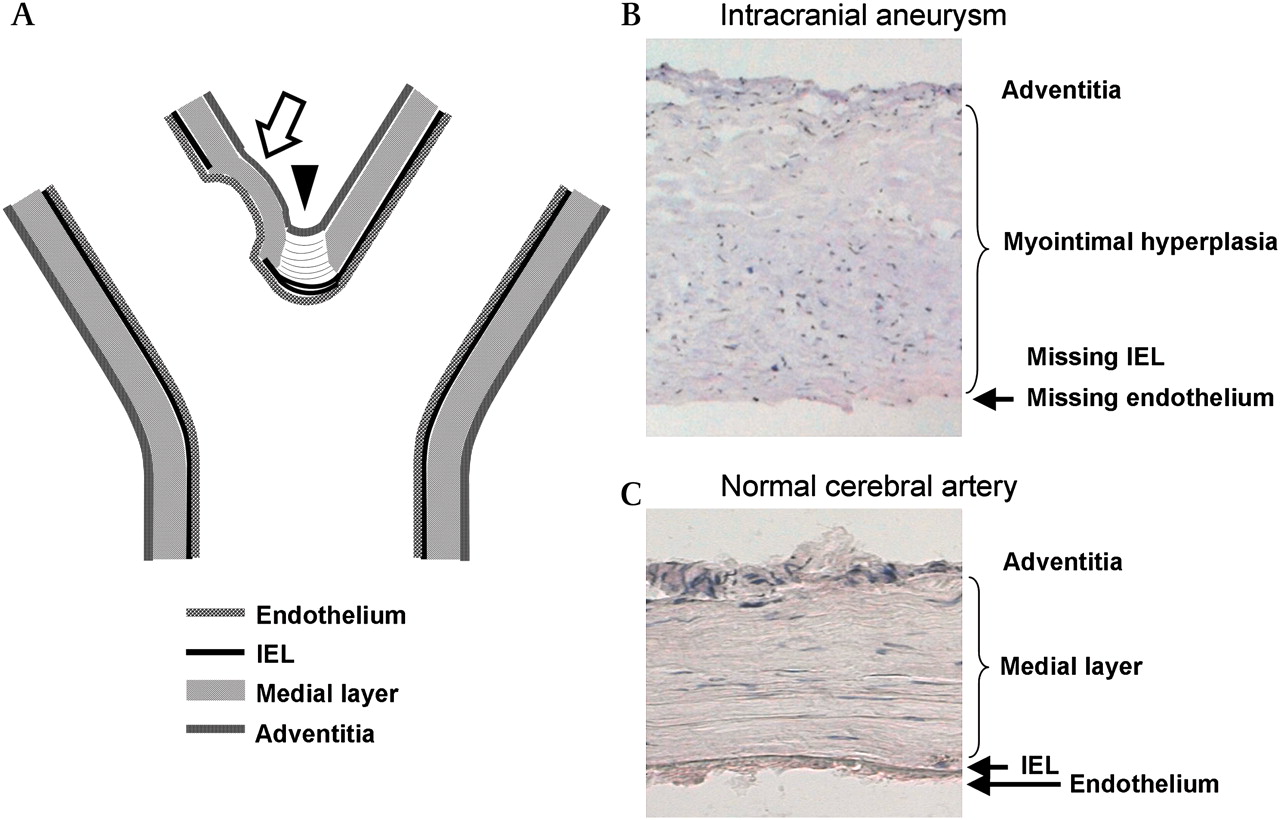
Intracranial aneurysm forms at the cerebral artery bifurcation. Often the initial nodus of the aneurysm (arrow) is located slightly distal to the fundus, as seen both in experimental animal studies and in human autopsy studies (A), at the point of highest shear stress. During early IA growth, the bifurcation also becomes involved. The branching point of the bifurcation (arrowhead; A) normally shows a gap in its medial layer (medial raphe), filled with tendon-like organized fibers, and the internal elastic lamina (IEL) shows some reduplication unrelated to aneurysm formation. The aneurysm wall lacks an IEL, and often shows disorganization within its wall (A, B). The normal intracranial artery wall (C) shows intact IEL, as well as linearly organized smooth muscle cells in its medial layer.
IA walls often show a continuum of degrees of degeneration
In histology, the IA wall often shows a transformation from almost intact to degenerated tissue in the neck-to-fundus direction. Degenerative changes in the IA wall include a decrease in the number of mural cells, these being mainly smooth muscle cells (SMCs), an excess of myointimal fibrous tissue and, ultimately, wall-thinning.15 16 Loss of SMCs is often especially prominent in the fundus, which may be almost lacking SMCs. However, the IA wall also undergoes a constant remodeling process, indicated by disorganization of wall structures, myointimal hyperplasia and SMC proliferation15 16 with associated activation of mitogen-activated protein kinases (MAPKs).17 18
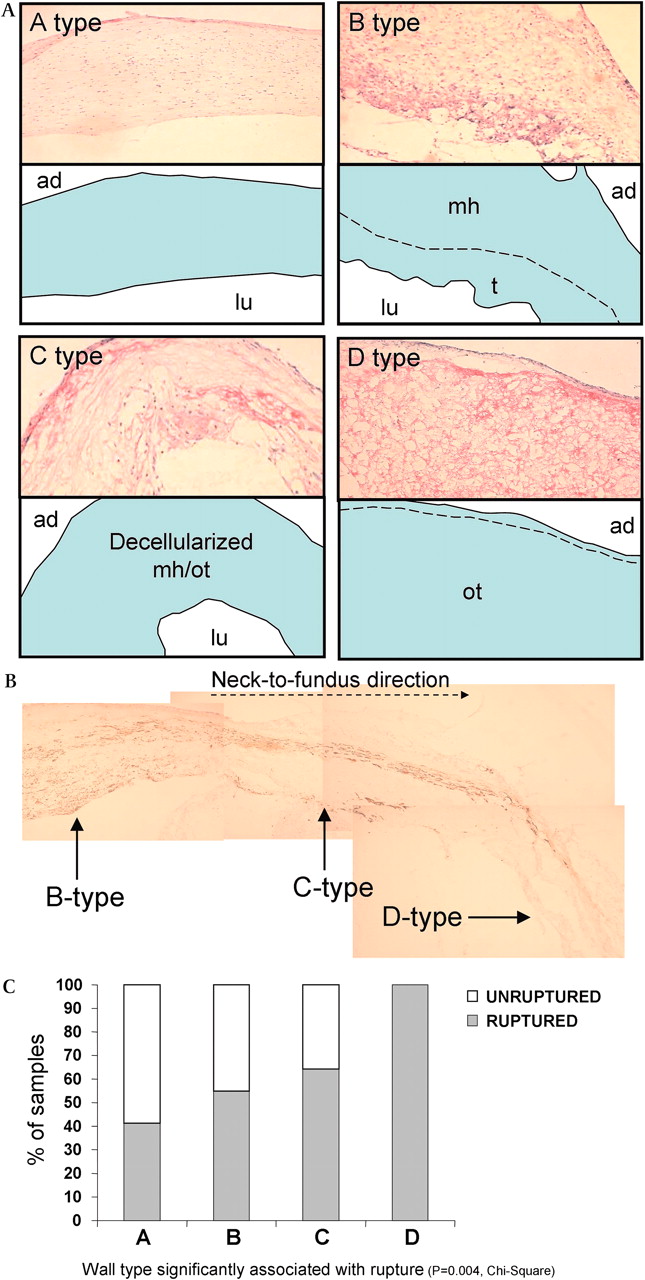
The histological changes are associated with IA's susceptibility to rupture.16 Frösen et al16 classified IA walls according to these parameters and found the lowest rupture risk within normal-looking IA walls, and a gradually increasing rupture risk for thicker walls with myointimal hyperplasia and an occasional thrombus, for thicker thrombosed walls with a reduced number of mural cells and for thin hypocellular walls with organized thrombi (figure 2). The study by Frösen et al16 reflects the significance of IA wall structure for its vulnerability to rupture.

Intracranial aneurysm (IA) wall structures and their relation to aneurysm rupture. IA walls often show varying wall structure with an increasing tendency toward rupture within the wall degeneration. Panel A represents different wall types classified as A, B, C and D types by Frösen et al16 and corresponding schematic graphs featuring histological characteristics of each wall type. The A type shows linearly organized smooth muscle cells and intact endothelium. The B type wall is thickened, with disorganized, proliferating smooth muscle cells (myointimal hyperplasia, mh) occasionally bearing a luminal fresh or organizing thrombus (t). The C type wall is thick but decellularized of former myointimal hyperplasia or organized thrombus (mh/ot). The D type wall is extremely thin and decellularized, with an organized luminal thrombus (ot). Photomicrographs of hematoxylin- and eosin-stained aneurysm samples show their typical histology. In the corresponding schemes, the aneurysm wall is in blue, and lu and ad stand for the luminal and adventitial side. Panel B shows a typical example of aneurysm wall degeneration and loss of smooth muscle cells (brown, myosin heavy chain) in the neck-to-fundus direction (dashed arrow). Panel C relates IA wall type to IA rupture.16 Figure 2A is modified from Frösen et al.59
IA wall degeneration in part due to cell death
A portion of the degenerative process of the IA wall is disappearance of cells, especially of SMCs.15 16 19 Extracellular matrix (ECM), synthesized mainly by SMCs (and possibly by myofibroblasts), maintains the wall's structural strength. In the normal cerebral artery wall, SMCs are mainly of the contractile type, enabling the fine regulation of blood flow. In IA, however, SMCs have undergone at least a partial phenotypic modulation from the contractile toward the synthetic type.20 The loss of matrix-synthesizing cells leads over a longer period to a decrease in synthesis and turnover of ECM and thus to structural weakness of the IA wall.
Both apoptosis and necrosis, two types of cell death, have been detected in the IA wall.16 19 21–24 In apoptosis, cells die in a preprogrammed manner as a response to death signaling resulting in a minimal proinflammatory effect. In contrast, necrotic cell death in response to an acute injury leads to a burst of highly proinflammatory cellular contents.
Inflammation and IA wall
Inflammatory cell infiltrations in IAs associate with fibrosis and IA wall degeneration
The majority of IAs (83–84%) rupture at the fundus, and the rest within the proximal part; only 2% of ruptures occur at the neck itself.25 26 Interestingly, inflammatory cell infiltrations have been detected in unruptured IAs in similar areas in the same proportions. In an autopsy series of 79 unruptured IAs, 29% harbored polymorphonuclear leucocytes and other (inflammatory) cells. In the majority of cases (78%), the cellular infiltrations occurred in the fundus and were always accompanied by fibrosis.26 More importantly, fibrosis never existed without an (inflammatory) cell infiltration, suggesting it is secondary to inflammatory cell invasion.26 Furthermore, in an autopsy series of 289 ruptured IAs, the margin of the rupture site always contained fibrin.26
Fibrosis is considered the end-phase of chronic inflammatory reactions.27 Crompton26 identified the inflammatory cells seen beneath the endothelium and deep in the IA wall as polymorphonuclear leucocytes, plasma cells and small round cells. In addition to polymorphonuclear leucocytes, immunohistochemical analyses have verified the inflammatory cells in IA walls to be mostly macrophages and T lymphocytes (T cells), some natural killer cells (NK cells) and a few B lymphocytes (B cells).10 15 16 28 The magnitude of inflammatory cell infiltration has been associated with IA rupture,15 16 underlining further the role of inflammation in the IA wall degenerative processes.
Unknown trigger of inflammatory reaction
An increase in macrophage, T cell and leucocyte infiltrations in the IA wall is associated with IA rupture,15 16 but the initial trigger for inflammation in IAs is unknown. Experimental models of hypertension-induced IAs in the rodent and canine imply that inflammatory cell infiltration is not necessarily the primary step in IA formation but manifests in the preformed IA at a later phase. In experimental IAs in rodents, macrophage infiltration follows IA formation and endothelial dysfunction29 30 as a response to chemotaxis from the experimental IA wall.31 Because in human IAs we cannot perform a similar analysis, the timing of inflammatory cell infiltration within the progress of human IA formation is unknown. In studies on early changes in IA formation, Stehbens32 found practically no inflammatory cells in early aneurysmal changes or areas of thinning, but detected small round cell infiltrations in some small IAs. However, it should be noted that in the 1960s, cell analysis was based on morphology alone. Moreover, unruptured and thus less degenerated IAs harbor fewer inflammatory cells.15 16 26
The role of endothelium and its hemodynamics in triggering inflammatory cells
Endothelial damage is one of the first changes in hypertension-induced experimental IA in rodents.29 The pressure and shear stress from circulating blood is necessary for normal endothelial cell function.33 As the flow alters during IA progression, the hemodynamics on the endothelium also change, probably sustaining molecular signaling of proinflammatory and proliferative pathways.33 As a probable sign of dysfunction, human IA endothelial cells express a monocyte chemoattractant protein-1 (MCP-1)31 34 35 highly chemotactic to inflammatory cells, macrophages, T cells, NK cells and basophils, and the vascular cell adhesion molecule-1 (VCAM-1)10 36 aiding in leucocyte recruitment. Their induction emerges probably through activation of transcription factor nuclear factor kappa B (NFκB), as seen in rodents in the early phase of experimentally induced IA.37 NFκB is also highly activated in human IA, mainly in the intima.37 NFκB is involved in cellular responses to stress, to cytokines, to free radicals and to oxidatively modified low density lipoprotein (oxLDL), and it plays a key role in regulating immune responses. In studies of experimentally induced IAs in rodents, MCP-1 was expressed in endothelium and accompanied by macrophage infiltration into the IA wall. Blocking of MCP-1 reduced the occurrence of these experimentally induced IAs.31 Also atherosclerotic lesions have increased endothelial expression of MCP-1.38 Interestingly, MCP-1, along with other chemokines, cytokines and growth factors, is an important regulator of fibrosis.27
Proinflammatory function of the complement system
The complement system is a biochemical cascade; it belongs to the innate immune system, because it becomes activated in response to pattern recognition and is not adaptable. Markers of complement activation have been evident consistently within IAs, both unruptured and ruptured, and in small amounts even in those IAs still lacking inflammatory cells.10 19 Interestingly, extension of complement activation also correlates with IA wall degeneration and rupture status, indicating the role of complement activation in enhancement of inflammation in the IA wall. Complement activation products, the anaphylatoxins C3a and C5a, are chemotactic to inflammatory cells and induce activation and degranulation of endothelial cells, mast cells and macrophages, thus indirectly inducing SMC contraction and capillary permeability. Complement activation may also induce apoptotic cell death.39 All these phenomena probably play a role in IA pathobiology.
The complement activation in IAs occurs through a classical pathway based solely on pattern recognition.54 Detectable in the IA wall are many of the potential complement activators capable of binding C1q and activating the complement: the immunoglobulins G (IgG) and M (IgM, comprising mainly naturally occurring antibodies), C reactive protein (CRP), apoptotic cells and oxLDLs.10 54
IA wall modulation and its relation to inflammation
The role of inflammation in IA wall remodeling
Along with degeneration, some IAs also show arterial remodeling, myointimal hyperplasia and SMC proliferation, as seen especially in ruptured IAs (40–48%).16 24 In part, the remodeling reaction may be stress induced rather than being a response to mitogenic stimuli, as indicated by upregulation of phosphorylated MAPK pJNK (c-Jun N-terminal kinase) in IAs of larger size and in ruptured IAs.17 Interestingly, both MCP-1 and complement may also contribute to this IA wall remodeling: MCP-1 inhibition reduces neointimal hyperplasia in experimental balloon-graft injury in rabbits,40 and in mice it reduces development of atherosclerotic changes manifested in the early phase by intimal hyperplasia and later by inflammatory cell infiltration and degenerative changes.41 Complement inhibition in rodents also restricts neointima formation and reduces the number of accompanying macrophages and T cells and progression of atherosclerotic changes.42 43 In human IAs, few intramural cells have been found to express MCP-1,31 similarly to experimental balloon-graft injury, which exposes an arterial wall to stretch, pressure and endothelial injury.44 An activated complement system indeed has been universally detectable in IAs.10 19 Whether these inflammatory mediators are primary or are secondary to the myointimal hyperplasia in IAs cannot be determined by data available thus far. However, experimental studies suggest the importance of inflammatory mediators in myointimal hyperplasia. In IA pathobiology, proinflammatory mediators may thus play a dual role.
Lipid accumulation and atherosclerotic changes in IAs: the ultimate manifestation of chronic inflammation
Atherosclerotic changes develop in the arterial wall as a reaction to mechanochemical stress. In atherosclerosis of peripheral arteries, traditionally the early lipid accumulation is accompanied by myointimal hyperplasia, complement activation, inflammatory cell infiltration and ultimately local accumulation of cholesterol and lipids in the arterial wall. This mature atherosclerotic plaque sometimes calcifies.45 The inflammatory response is believed to be initiated by intimal accumulation of LDL further modified into proinflammatory oxLDL by enzymes and oxygen radicals. The ensuing inflammatory response is mainly mediated by complement activation, several cytokines, growth factors and other mediators secreted by inflammatory cells. Of these, macrophages and T cells are believed to determine the fate of the nascent plaque depending on the balance between proinflammatory and anti-inflammatory responses.46 Hemodynamic factors may assist in the LDL accumulation, because increased shear stress upregulates the LDL-receptor expression of endothelial cells.47
The role of atherosclerosis in the etiology of IA has been under debate for decades. If the classification of atherosclerosis by histological changes is strictly followed, nearly all IAs can be considered atherosclerotic lesions.28 Advanced atherosclerotic modifications of IAs, featuring a core of atheromatous debris encapsulated by fibrous tissue accompanied by macrophages and T cells, have been evident in approximately half of all IAs,28 48 49 whereas myointimal hyperplasia occurs in the other half.28 Lipids and LDLs accumulate in the intima of normal cerebral artery walls during the life course,50–52 with LDLs detected immunohistochemically, especially in the endothelial cells of cerebral artery bifurcations and also within the ECM associated with intimal thickening.52 Lipids and oxLDLs accumulate in the unruptured IA walls accompanying an activated complement system.53 54 However, whether such lipid accumulation should be considered as a preliminary, proinflammatory state in IA pathobiology or as secondary to initial IA formation needs further study.
Several growth factors contribute to IA wall remodeling
In part, proliferation of mural cells of the IA wall, mainly the SMCs, may occur as a response to several growth factors detected in the wall. Growth factors stimulate inflammatory cells, but on the other hand, inflammatory cells are in part responsible for the production of growth factors. Macrophages are an especially important source of growth factors and cytokines that affect the fate of SMCs and fibroblasts.55 Platelet-derived growth factor, basic fibroblast growth factor (bFGF) and transforming growth factors α (TGF-α) and β (TGF-β) are expressed in IAs in immunohistochemistry56 57 and at varying transcriptional levels.35 36 Also strongly expressed in IA walls is vascular endothelial growth factor (VEGF), which functions in angiogenesis, vasodilatation and chemotaxis of inflammatory cells.56 58
The mural cells of the IA wall express receptors for many growth factors. Receptor-1 for bFGF is associated with minor leaks, and receptors for TGF-β are associated with IA rupture (TGF-βR2)35 59 and wall remodeling (TGF-βR3).59 Expression of VEGF receptors is associated with IA wall rupture, remodeling, T cell and macrophage infiltration (VEGF receptor-1), and proliferation and myointimal hyperplasia (VEGF receptor-2).59 In addition, IAs also express receptors for insulin-like growth factor (IGF), which promotes cell proliferation.59 Based on expression of growth factors and growth factor receptors in IAs, the IA wall seems to undergo a constant remodeling process,59 probably in response to inflammation and local stress, as indicated by differences in its pJNK expression.17
The role of macrophages and lymphocytes
What role for acquired immunity?
IgG is also a potent activator of the complement system, and both are detectable in IAs.10 19 IgG is produced mainly by prior immunization for a specific antigen and activation of acquired immunity. Triggering of the acquired immune response and production of IgG-class antibodies targeted against specific antigens necessitates prior expression of the antigen to an adjacent T cell. Depending on antigen origin, it is expressed in a class I or II major histocompability complex (MHC-I or MHC-II) surface protein. T lymphocytes have been regularly detected in immunohistochemical studies of the IA wall10 24 28 and are associated with IA rupture.15 16 Furthermore, at least some of the ‘small round cells’ noted in early histomorphological studies of the IA11 12 26 32 have likely been lymphocytes. Interestingly, in histology, macrophages expressing MHC-II have been located in close contact with activated CD45RO-positive T cells in IA walls28 indicating an active macrophage-aided T cell response. Occasionally, the scattered SMCs within advanced atherosclerotic lesions in IAs have also expressed MHC-II.28 35 In gene expression analysis, the antigen presentation pathways and MHC-I and II expression have been upregulated in most pathway clusters.35 In addition to T cells, a few B cells, required for immunoglobulin production, have been detectable in IAs.10 Whether or not a specific antigen-mediated immunoreaction against IA wall structures plays a role in IA wall inflammation warrants further study.
Cytokines directing cellular responses of IA wall: roles of Th1 and Th2
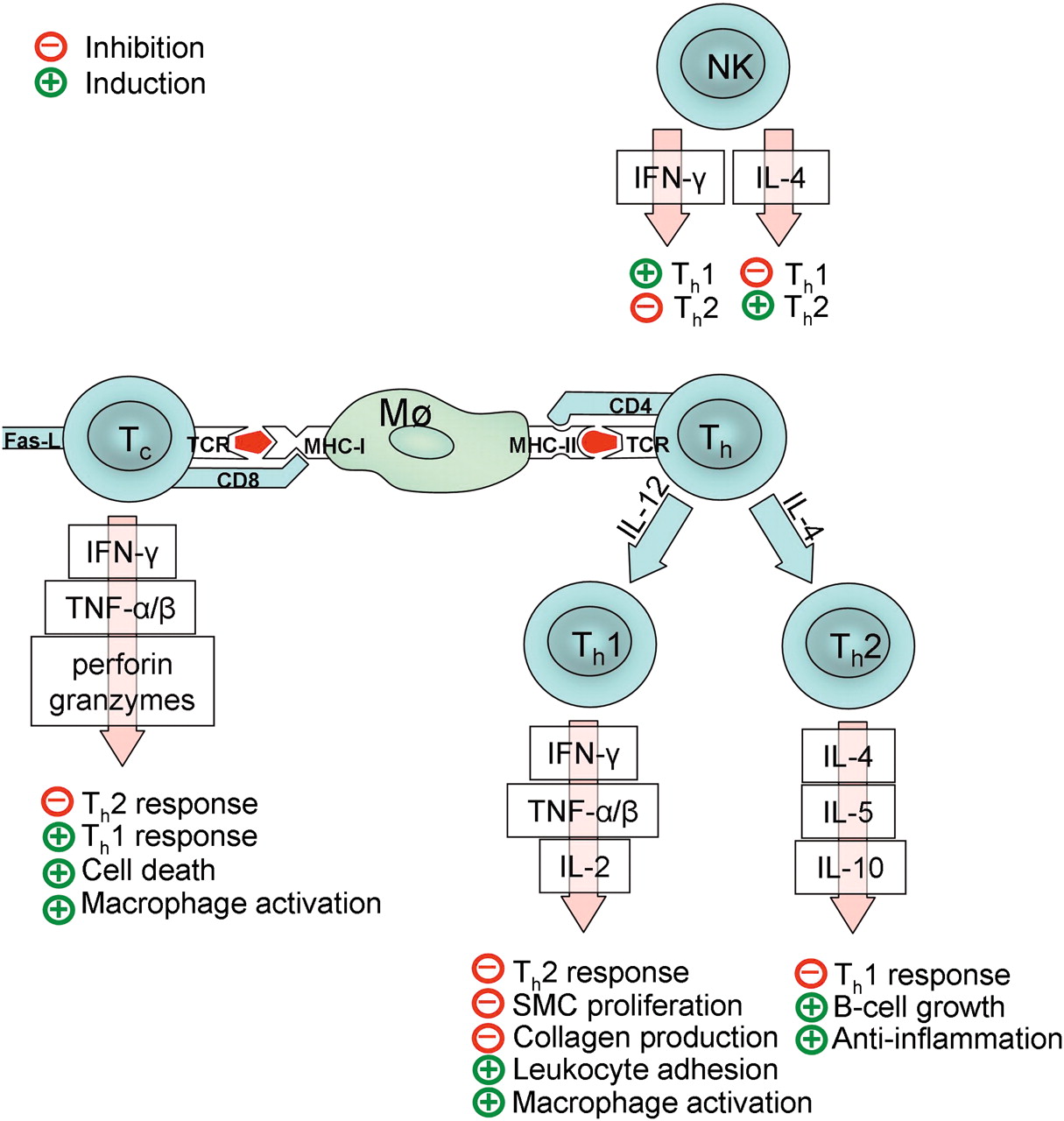
The function of T cells depends on their phenotype: cytotoxic CD8-positive T cells (Tc cells) secrete the proinflammatory cytokines interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) and may also induce apoptosis by Fas-ligand-dependent interaction. The actions of CD4-positive helper T cells (Th cells) depend on their differentiation in either the Th1 or Th2 direction, directed by the presence of interleukin (IL) 12 or IL-4. Th1 and Th2 cells differ in the profile of cytokines they produce: Th1 cells produce mainly IFN-γ, TNF-α, TNF-β and IL-18, which activate macrophages and NK cells, whereas Th2 cells produce mainly anti-inflammatory IL-4, IL-5 and IL-10, which induce B cell growth and anti-inflammatory effects. The activation and polarization of the adaptive immune response is summarized in figure 3.
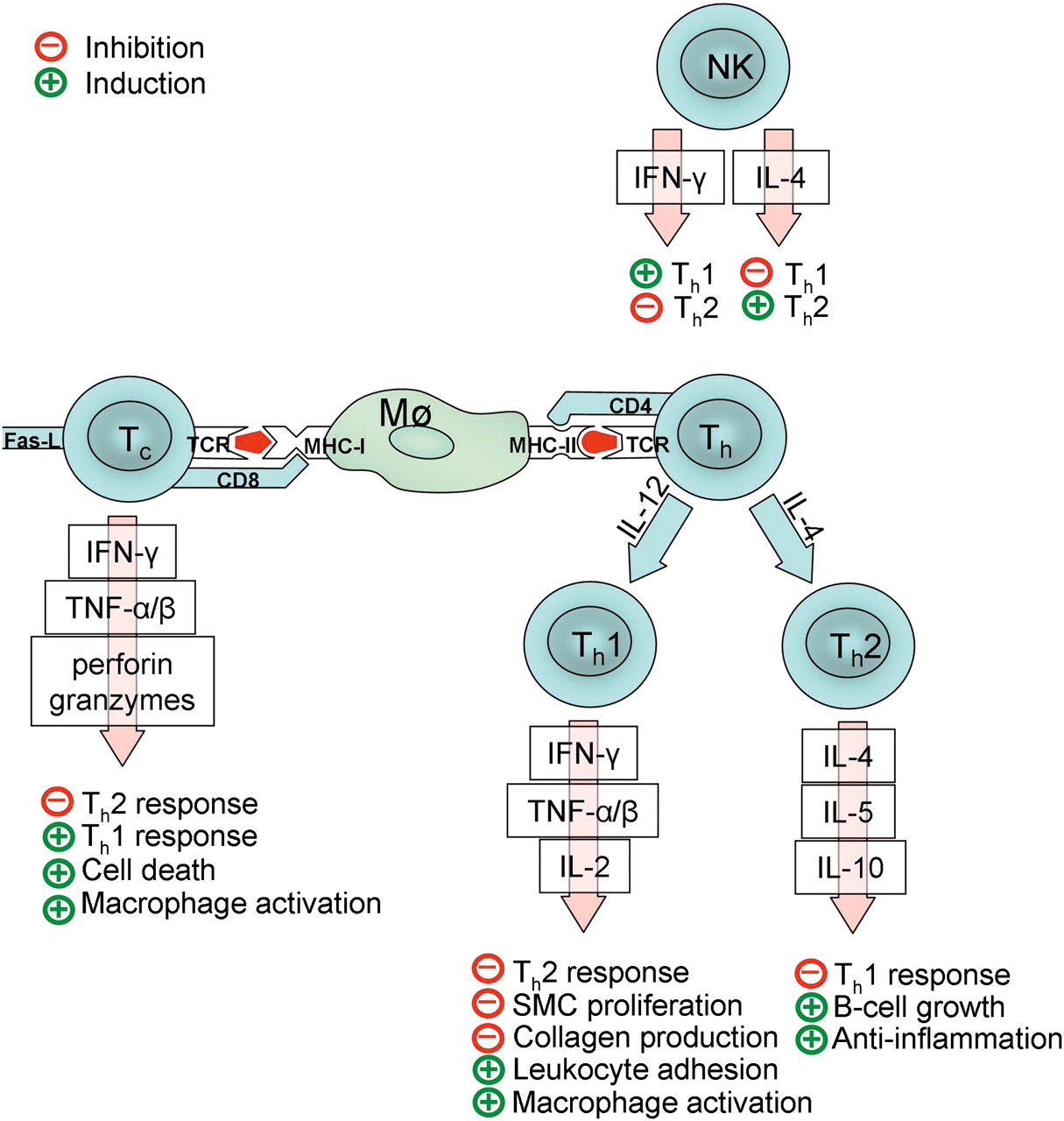
Simplified diagram of probable activation mechanisms and functions of adaptive immunity in intracranial aneurysms. A macrophage (Mø) or other antigen-presenting cell presents antigens (red) in a specific groove of major histocompability complex I or II (MHC-I or MCH-II) depending on antigen origin. A T cell recognizes the antigen through a T cell receptor (TCR), and the MHC molecule through CD4 (helper T cells, Th) or CD8 (cytotoxic T cells, Tc). Activated T cells secrete many cytokines and inflammatory mediators (interferon gamma, IFN-γ; tumor necrosis factor alpha and beta, TNF-α and TNF-β; interleukins, IL) that further guide the inflammation in the aneurysm wall. The polarization of Th cells in either the Th1 or Th2 direction depends on the presence of IL-12 or IL-4. NK cells play an important role in this process, as they may guide the polarization by secreting either IFN-γ or IL-4. Note that T cell regulation is a complicated process, with only its most important mediators presented here. Thus far, other important T cell types such as Th17 and Th3 cells and regulatory T cells have also been identified, but are excluded because their biological functions are still in most part unresolved.
The T cell profile in human IAs has not been verified for the present, although the lack of IL-1060 in ruptured IAs and the presence of TNF-α61 indicate either the dominance of the Th1 direction or a cytotoxic T cell response. Jayaraman et al60 report also finding Th2-type cells, but suggests that they are being suppressed and thus inactive. In atherosclerosis, the Th1-type cell response dominates.46 IFN-γ, produced by Th1 cells, Tc cells and activated macrophages, inhibits both SMC proliferation and collagen production and, together with IL-1β and TNF-α, induces the expression of several types of leucocyte adhesion molecules. At transcriptional level, receptors for IFN-γ are upregulated in IA walls.35 36
As the cytokines of the Th1 response inhibit the Th2 response and vice versa, the balance between Th1 and Th2 would have a substantial effect on the fate of IA regarding the proinflammatory and SMC-antiproliferative effects of the Th1response versus the anti-inflammatory alignment of the Th2 response. NK cells differ from T cells, because they produce cytokines in response to antigen presented in the CD1 molecule. The main cytokines produced by NK cells are IL-4 and IFN-γ, and thus an NK cell can proceed effectively in directing the T cell response in either the Th1 or the Th2 direction. NK cells have been detected in the human IA.28 Switching of the T cell response from a Th1 to a Th2 direction may induce progression of aneurysmal dilatation of an atherosclerotic aorta.62
Macrophage activation induces further IA wall inflammation and fibrosis
In the hypertension-induced experimental IAs in rodents, macrophages have been the first population of inflammatory cells infiltrating into a preforming IA, secondary to endothelial dysfunction.29 Macrophages weaken the IA wall by secreting ECM-degrading proteolytic enzymes and inducing the apoptotic death of SMCs. They also induce fibrosis through secretion of TGF-β and produce numerous other effectors, that is, reactive oxygen species (ROS), tumor necrosis factor α (TNF-α) and IL-1, as reviewed by Boyle.55 The multiple potential effects of macrophage activation on the IA wall are summarized in figure 4.
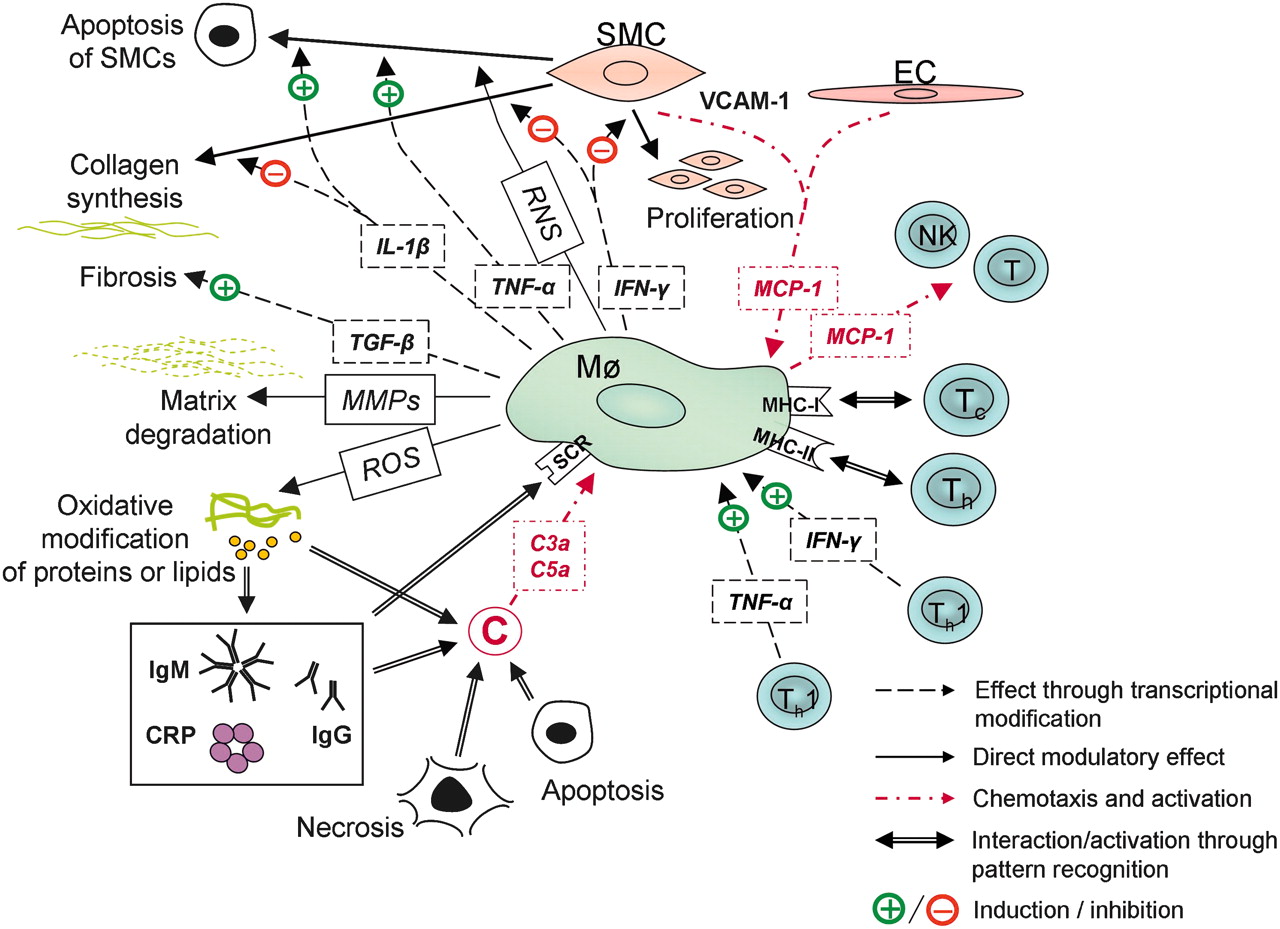
Probable activators and main functions of macrophages in intracranial aneurysms. C, complement system; C3a and C5a, anaphylatoxins; CRP, C reactive protein; EC, endothelial cell; IFN-γ, interferon gamma; IgG, immunoglobulin G; IgM, immunoglobulin M; IL-1β, interleukin 1-beta; Mø, macrophage; MCP-1, monocyte chemotactic protein; MHC-I and MHC-II, major histocompability complexes I and I; MMP, matrix metalloproteinase; NK, natural killer cell; RNS, reactive nitrogen species; ROS, reactive oxygen species; SCR, scavenger receptor; SMC, smooth muscle cell; T, T cell; TGF-β, tissue growth factor beta; TNF-α, tumor necrosis factor-alpha; VCAM-1, vascular cell adhesion molecule-1.
TNF-α61 occurs in the human IA and the expression of receptors of TGF-β is associated with IA wall remodeling and rupture.59 Disruption of the IL-1β gene in mice leads to a lower percentage of advanced experimentally induced IAs and apoptotic cells in these IAs.63 In cultured rat and human aortic SMCs, IL-1β reduces, through activation of NFκB, the biosynthesis of collagens I and III and of the lysyl oxidase, needed for collagen cross-linking. In the rat, during progression of experimental IA, lysyl oxidase expression is reduced first in the medial SMC, followed by reduction in the adventitia.64 The effect of IL-1β on collagen synthesis of human arterial SMCs is similar.65
In the human IA, TNF-α expression is related to expression of induction of the proapoptotic pathway.61 Although TNF-α is a proinflammatory cytokine, it may also play some protective role, because in the arteries of TNF-receptor-deficient mice, atherosclerotic modifications are accelerated.66 Induction of TNF-α is associated with many IA risk factors, specifically hypertension, age, female gender, smoking and alcohol abuse.60 In part, TNF-α may act through activation of other cytokines or signaling molecules (eg, IL-1 release, activation of matrix metalloproteinases (MMPs) and of endothelium). In IA pathobiology, the multiple effects of macrophages on IA wall matrix, mural cells and other inflammatory cells make macrophages extremely potent cells.
ROS can modify IA structures to become proinflammatory
In experimental IA studies, ROS produced by infiltrating macrophages and SMCs play an important role in IA growth.67 ROS can modify the proteins and lipids of a wide variety of extracellular and cellular components.68 The oxidized phospholipids as well as oxidized lipoproteins formed are proinflammatory, since they are recognized by natural antibodies,69 innate soluble proteins like CRP70 and complement C1q71 72 and (macrophage) scavenger receptors.73 OxLDLs as well as IgM (the main subtype of natural antibodies), CRP, C1q,54 and macrophages (expressing scavanger receptors),16 have been detectable in the IA wall suggesting the active function of ROS.
Free hemosiderin, an iron-storage complex of erythrocytes, and even intact erythrocytes, have appeared in the IA wall,15 24 74–77 probably indicating silent intramural leakage. The possible source for leakages can be neovessels visible in IA walls, especially in areas showing atheromatous changes in wall structure.26 ROS may dissociate heme from its protein-bound state, creating pro-oxidative free heme and free iron ions.78 Similarly to ROS, reactive nitrogen species originating from nitric oxide (NO) also possess an oxidative capacity. In inflammation, activated macrophages produce excess amounts of NO, leading to a relative lack of antioxidants and to formation of free radicals.55 In addition to macrophages, endothelial cells are another important source of NO, produced normally for vasodilatory and antithrombotic functions.79
MMPs reduce IA wall tensile strength
The tensile strength of the IA wall is provided by thin sublayers of collagen of mainly types I and III.80 81 Aneurysm wall remodeling and degeneration is evident in ruptured IAs along with increased macrophage infiltration.16 The factor responsible for critical disruption of IA wall mechanical strength and rupture is believed to involve MMPs. These are proteolytic enzymes normally needed for vascular remodeling, cellular migration, and processing of ECM proteins to provide vascular homeostasis. MMP activity is upregulated in response to hemodynamic factors, mechanical strain, vascular injuries, inflammatory cytokines and ROS. MMPs also play a role in neointimal formation.82 In IA patients, MMP-283 is responsible for increased serum gelatinase activity.84 The expression of membrane-type 1 MMP (MT1-MMP), and the gelatinases MMP-2 and -9 are increased85–87 in IA walls, especially in ruptured ones,17 87 indicating their role in IA wall structural weakening.
Types of MMP expression among IAs differ: MMP-2 is expressed in most IAs, whereas MMP-9 is especially expressed in IAs with atherosclerotic changes.85 86 In immunohistochemical staining, MMPs are expressed by mural cells that are mainly SMCs.86 In addition to SMCs, macrophages serve as an important source of MMPs. MMP-2 and MMP-9 appear also in experimental IAs in relation to macrophage infiltration.30 MMP inhibition does not, however, prevent experimentally induced aneurysmal changes in rats,30 88 although the incidence of experimentally induced advanced IAs is reduced.30
In addition to proteolytic activity, MMPs may have other functions in IAs as well. MMP-2 and MMP-9 inhibit rat SMC contraction, MT1-MMP and MMP-2 enhance angiogenesis, and degradation of ECM by MMPs releases several angiogenic growth factors.82 Other proteolytic enzymes may also play a role in IA wall structural weakening, since the expression of cathepsin D by mural SMCs and by macrophages and of cathepsin G by leucocytes, is increased in ruptured and thus furthermore degenerated IAs.15 Other cathepsins, S, B and K, are expressed in macrophages, endothelial cells and SMCs in human IAs.89 In experimental IA models, expression of cathepsins is higher.89
MMPs are secreted in inactive proforms activated by protein cleavage by other proteinases, those secreted, for example, by macrophages.55 In an IA, TNF-α60 and NO may in part be responsible for MMP activation. Other important endogenous regulators of MMPs are endogenous tissue inhibitors of metalloproteinases (TIMPs), which reduce the excessive proteolytic activity of MMPs.82 In experimentally induced IAs in rodents, TIMP-1 and TIMP-2 levels increase in the early stage of IA formation, but soon decrease, promoting progression of the experimentally induced IA.90 TIMPs are expressed mainly by SMCs. Similarly, in the human IAs TIMP levels are decreased, potentiating the effects of increased MMP levels.60 87 Cathepsins also have their own endogenous inhibitors, for example, cystatin C for cysteine cathepsins S, B and K. Along with an increase in cathepsin expression, human and experimental IAs show a complementary decrease in cystatin C expression.89 Thus, for IA growth, the balance between activation of proteolytic enzymes and their inhibition seems critical.
Matrix failure and IA rupture
Long-term imbalance of matrix synthesis and degradation weakens the IA wall
The IA, as with other long-term chronic inflammatory states, shows an accumulation of fibrotic tissue and related cell death. Fibrosis develops in the responses of SMCs and myofibroblasts to cytokines and growth factors secreted by inflammatory and other activated cells. Myofibroblasts, differentiated probably from endothelial or circulating progenitor cells or activated fibroblasts, are reparative α smooth-muscle actin-positive cells that may also express other SMC phenotype markers, depending on their level of differentiation. Transcription of procollagens I and III is promoted by TGF-β, secreted in large part by macrophages.27 Polarization of an acquired immune response also affects fibrotization: IFN- γ and IL-12, both secreted by Th1-type lymphocytes, are antifibrotic, whereas the Th2-type immune response upregulates the fibrosis related to healing of tissue trauma. Autoantibodies and IL-6 secreted by B cells are known to activate myofibroblasts (reviewed by Wynn27). In the human IA, a Th1-directed immune response may dominate that of Th260 like that seen in atherosclerosis.91 This would promote a relative decrease in the synthesis of structural proteins.
During IA progression, the change in SMC phenotype from contractile to synthetic and the increase in relative numbers of SMCs (increased SMC proliferation and myointimal hyperplasia), may compensate for the increased MMP expression and sustain IA wall strength. In concordance with this theory, the IA wall shows, in in situ hybridization, increased synthesis of collagen type III despite a slight diminution in expression of collagen type III in immunostaining when compared with the normal control artery, probably indicating faster collagen turnover.81 However, as the mural cells gradually die during IA progression, their ECM-synthesizing capability decreases, as does the matrix turnover, leading to a decrease in net tensile wall strength and an increase in susceptibility of the IA to rupture.
Cell death is induced by an inflammatory response
Inflammation is a proapoptotic state, and inflammation involves several potential mechanisms that may induce apoptosis also in the IA wall. Many cytokines secreted by inflammatory cells are proapoptotic, and oxidative, hypoxic and other kinds of stress may lead to activation of an intrinsic apoptotic pathway. Tc cells and NK cells secrete granzymes, perforin and ganulysin. Granzymes, being serine proteases, lead to apoptosis by activation of either a caspase (especially caspase-3) or a mitochondrial pathway. Perforin forms a Ca2+ ion-conducting pore on a target cell membrane, leading to cell lysis and necrosis unless the cell manages to endocytose the pore programming apoptotic death. Granulysin may act through caspase-3 activation or in a cytolytic manner, being also chemotactic to other inflammatory cells.92 Like perforin, the pore-forming terminal complement complex C5b-9 may, in large amounts, lead to cell lysis and necrotic cell death, but induce apoptotic cell death in sublytic concentrations.39 Terminal complement complexes are detectable in human IAs, especially in decellularized areas and because of a lack of protectin, that inhibits C5b-9 formation.19 54 Apoptotic cell death may also be induced by direct contact between activated T cells or NK cells and target cells through Fas–Fas-ligand recognition. This launches an intracellular signaling cascade leading to caspase activation and the apoptosis of the target cell.92 93 TNF-α, produced by macrophages, elevates activation of the apoptotic, Fas-associated death domain protein pathway, shown also in the IA.61 SMC detachment from surrounding ECM structures by proteolytic enzymes may induce apoptotic cell death, called in this context anoikis.94 Except for the presence of Fas-associated death domain protein activation61 and caspase-323 in the IA, the existence of neither of these proposed pathways leading to cellular death has been evident in IA walls.
Thrombosis
The presence of an organizing thrombus is associated with IA rupture.16 Normally, intact and healthy endothelium protects the luminal surface of the vascular wall from thrombosis and platelet aggregation by expression of CD39 and secretion of NO and prostacyclin.95 Endothelial dysfunction is related to IA formation in experimental models,29 and partial or total de-endotheliazation is associated with human IA rupture.15 16 Damaged or stressed endothelial cells release inflammatory mediators that initiate an antifibrinolytic coagulation cascade.27 In total endothelial injury, the subendothelial prothrombic structures that come into contact with platelets facilitate the activation of the coagulation cascade and thrombus formation. In addition to probable ongoing inflammation of the IA wall, altered hemodynamics and decreased blood flow also promote endothelial dysfunction and thrombus formation.96
Both erythrocytes and platelets become trapped in the fibrin meshwork of the fresh thrombus, and chemotaxis and adhesion-molecule expression aid the infiltration of neutrophils and macrophages into the injury site.27 96 Erythrocytes start to lyse within 2 days, releasing intracellular components that induce oxidative stress and activate complement.97 To promote fibrinolysis and thrombus deformation, platelets and neutrophils secrete MMPs, cathepsin G and elastase. The proteolytic enzymes released by neutrophils and macrophages are probably involved in further degradation of the IA wall as well as in induction of anoikis.98 99 Later, the inflammatory cells may migrate into the IA wall; polymorphonuclear leucocytes and other inflammatory cells have been evident in IA wall in histology.12 26 Thrombi start to organize by invading cells, SMCs or myofibroblasts that originate at least in part in the vascular wall,27 100 101 then becoming an indistinguishable part of the IA wall.
Summary of inflammatory changes in the IA wall
In summary, chronic inflammation seems to have multiple functions in the IA wall, favoring both IA wall degeneration and reparative mechanisms. However, if the net effect is degenerative, inflammation leads to critical thinning of the IA wall and ultimate rupture (figure 5). In unruptured IAs, what is unknown is whether the inflammatory degenerative mechanisms are totally compensated to become the steady state, or the IA wall degeneration is still proceeding undetectably slowly. Combining data from experimental IA studies in animals and studies of human IAs reveals that an IA seems to form and grow in response to hemodynamic stress and a proinflammatory milieu that recruit inflammatory cells, exacerbated by endothelial dysfunction. Probably as a compensatory mechanism for IA formation, SMCs proceed to proliferation, aided by the presence of several growth factors. However, the increased expression of cytokines, oxidative stress, complement activation and ECM-degrading proteolytic enzymes originating from inflammatory cells and proliferating SMCs, promotes fragmentation of IA wall structures and induces cell death. Modulation of IA wall structures may also lead to formation of new immunogenes that further trigger inflammation. In addition, some IAs become thrombosed, which further amplifies the ongoing inflammatory reaction and wall degeneration. With the net effect of cell death and decreased matrix turnover, the IA wall gradually degenerates, loses its tensile strength and ultimately ruptures.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hypothetical representation of intracranial aneurysm wall degeneration and rupture. An aneurysm forms at the site of disrupted internal elastic lamina (IEL) (1) associated with myointimal hyperplasia (2). Hemodynamic stress, primary or secondary to the structural modifications of the vascular wall, leads to endothelial dysfunction (3) and to chemotaxis for inflammatory cell invasion (4). At the same time, matrix synthesis is increased to support the tensile strength of the aneurysm wall due to the function of growth factors secreted, for instance, by macrophages (5). However, due to the cytotoxic milieu caused by inflammatory cells, by the increased matrix and possibly by decreased oxygen supply due to increased wall thickness, the mural cells (mostly smooth muscle cells, SMCs) begin to die (6). Matrix turnover slows due to its decreased production secondary to decellularization and to increased lysis by the action of matrix metalloproteinases (7). Over time, the aneurysm wall becomes mechanically more fragile, and when intraluminal pressure overcomes tensile strength, it finally ruptures. Lymphocytes also play an important role in aneurysm wall degeneration by guiding the immune response and the surveillance of mural cells. However, their timing during aneurysm progression is unclear (X). Depending on hemodynamics and shear stress, the endothelium undergoes severe dysfunction or may die, either process leading to thrombus formation (8). In addition to other inflammatory cells, the thrombus attracts neutrophils (9), potent producers of proteolytic enzymes. Over time, the thrombus may become organized by migrating mural cells (10). However, due to cell death and decreased matrix turnover, eventually the aneurysm wall becomes sufficiently fragile to rupture. This figure is a simplification of the pathobiological processes within the aneurysm wall and thus cannot illustrate other factors probably playing crucial roles in aneurysm wall degeneration, such as complement activation, lipid accumulation and neovascularization.
Future perspectives
Imaging inflammation
Unruptured IAs may remain unchanged over years, and even be unlikely to rupture at all.102 In order to direct prophylactic but invasive treatment to those IAs at high risk of rupture, they must be identified. As the presence of inflammation and other histopathological features correlates with the probability of IA rupture, the ability to image inflammation in unruptured IAs could aid in clinical decision-making concerning IA closure and rupture prevention.
Data on imaging inflammation in human IAs have not, to our knowledge, been reported. However, DeLeo et al103 managed to image bacterial lipopolysaccahride-induced inflammation in the elastase-induced rabbit experimental IA. They were able to detect experimental IA wall macrophages and neutrophils by use of a myeloperoxidase-specific magnetic resonance contrast agent, useful in visualizing mouse myocardial ischemia-reperfusion injury104 as well as rabbit atherosclerotic plaque.105 Imaging of vascular inflammation has been studied in humans principally within the atherosclerotic plaques and vasculitides. Radiolabeled [18F]fluorodeoxyglucose in positron-emission tomography recognizes metabolically active macrophages and distinguishes between vulnerable and stable plaques.106 Signal intensity correlates with activated CD68-positive macrophage density107 in carotid plaques as well as in abdominal aortic aneurysms.108 Macrophage phagocytic activity can be detected and verified by ultra-small superparamagnetic iron oxide particles and imaged with T2-weighted magnetic resonance sequences, as performed for human carotid plaques.109 110 Radiolabeled Annexin V detects phosphatidylserines on the apoptotic cell and has been successful in imaging by single photon emission tomography the extent of apoptosis in experimental aortic atherosclerotic plaques in rabbit111 as well as in human carotid arteries.112
New treatment methods
Currently the only IA treatment methods are clipping or endovascular therapy by coils or other embolization materials. These treatment methods are, however, both invasive and somewhat risky.3 For those patients in need of closure of their unruptured IA susceptible to rupture or of their already ruptured IA, improvement in coil technology such as coated coils may allow even more effective and permanent treatment results. Thus far, coils releasing growth factors have been tested in animal models with varying results. Local release of bFGF induces proliferation in the arterial wall of the rat common carotid artery,113 includes fibrosis and obliteration of rabbit venous-pouch and elastase-induced aneurysms,114 115 and causes improvement in occlusion of ligated common carotid arteries in the canine.116 Local release of VEGF enhances, in the rat, vascular wall thickness, endothelial formation, cell infiltration, lumen reduction and fibrotization of ligated common carotid arteries.117 118 TGF-β, however, has led to no improvement in long-term occlusion of rabbit elastase-induced aneurysms119 nor in the canine model.120 It is, however, noteworthy that although the surgically created aneurysm models commonly used in the testing of different embolization materials may resemble human IAs in structure, their inflammatory milieu may differ. Moreover, because many human IAs are extensively decellularized, the basis for growth factor-aided occlusion of the aneurysmal sack differs from that in animal models. The efficiency of such a treatment thus warrants thorough further studies.
The option of systemic pharmaceutical treatment
As inflammation can be controlled by several pharmaceuticals, one intriguing idea is to halt progressive IA wall degeneration and turn it toward the steady state by pharmaceutical therapy, thus preventing IA rupture. Thus far, few experimental animal studies involve the effect of systemic pharmaceutical treatment on IA incidence and progression, many of them by Aoki et al121 122 in an experimental IA model in the rat. Some statins (simvastatin and pitavastatin), HMG-CoA-reductase inhibitors, have prevented IA progression (growth), wall degeneration (thinning) and also MCP-1, VCAM-1, IL-1β, and MMP-2 and MMP-9 expression, probably through inhibition of NFκB activation. Doxycycline, however, a nonspecific MMP inhibitor, fails, in rats, to affect the rate of experimental IA formation.88 Edavarone, a synthetic free-radical scavenger, inhibits experimental IA formation and reduces MCP-1, VCAM-1 and MMP-2 expression.67 Nifedipine, a calcium antagonist, inhibits the DNA binding of NFκB, leading to the reduced size and increased wall thickness of the experimental IA as well as preventing progression of IA wall degeneration and preventing enlargement of pre-existing IAs. MCP-1 and MMP-2 levels are also reduced.123 Successful pharmaceutical treatment options can reduce chemotactic and proinflammatory signaling, which would lead to an associated decrease in macrophage infiltration. However, none of the therapies has been efficient enough to prevent IA formation entirely. Since IA wall degeneration and inflammation involved in its processes are both complicated, the possible targets for treatment are many. Although the biology of human IA progression may be more complicated than is the progression of an experimental IA, results are somewhat promising and offer motivation for targeting research on anti-inflammatory treatment.
In conclusion
Despite all the research on IAs, the pathobiology behind IA formation and the exact mechanisms of IA rupture remain unknown. All the evidence gathered from human IA samples and from experimental studies of IA do point to the major role of inflammation in IA pathobiology. Studies on human IA samples have, however, been limited in locating the histopathological changes and comparing them with clinical parameters and IA rupture status. The only knowledge of phenomena related to IA formation comes from experimental studies in animals. Moreover, new treatment options have been tested in experimental IA alone. Because human IA may include additional pathobiological mechanisms for IA formation, growth, rupture or both, caution is necessary when extrapolating from animal findings. Development of finer imaging and more sophisticated endovascular treatment methods do, however, provide great possibilities for future IA diagnostics and treatment.
Acknowledgments
We thank neuropathologist Anders Paetau, MD, PhD (Dept of Pathology, University of Helsinki, Finland) for his expert comments, and Carol Norris, PhD (University of Helsinki) for professional language revision.
References
Footnotes
Funding This work was supported by Helsinki University Central Hospital (HUCH) EVO grant TYH 2009303.
Competing interests None.
Ethics approval This study was conducted with the approval of the HUCH Ethics Committee.
Provenance and peer review Commissioned; not externally peer reviewed.