Article Text

Abstract
OBJECTIVES The most common neurological manifestations in Wilson’s disease are parkinsonism and dystonia. These are assumed to be due to striatal injury, which has been repeatedly demonstrated by pathology and CT or MRI. The substantia nigra has not been shown to be damaged in pathological studies. However, there have been clinical and imaging studies suggesting presynaptic nigrostriatal injury. (1r)-2β-carbomethoxy-3β-(4-iodophenyl)tropane (β-CIT) is a specific ligand that binds to the dopamine transporter (DAT), and can examine the integrity of dopaminergic nerve terminals. Evidence for presynaptic nigrostriatal dopaminergic damage in Wilson’s disease was searched for using [123I]-β-CIT SPECT.
METHODS Six patients with Wilson’s disease were studied, together with 15 healthy normal controls, and six patients with Parkinson’s disease. After injection of [123I]-β-CIT, SPECT studies were done at 18 hours. Specific striatal/occipital binding ratio (S/O ratio) was calculated as (striatal binding−occipital binding)/occipital binding.
RESULTS The specific S/O ratios were 6.22 (1.32) (mean (SD)) in normal volunteers, 3.78 (0.65) in Parkinson’s disease, and 3.60 (0.49) in Wilson’s disease.
CONCLUSION There was severe loss of the DAT in the striatum suggesting significant damage in presynaptic nigrostriatal dopaminergic nerve terminals. Therefore, a presynaptic lesion may contribute to neurological manifestations in Wilson’s disease.
- Wilson’s disease
- dopamine transporter
- substantia nigra
- striatum
- nuclear imaging
Statistics from Altmetric.com
Wilson’s disease is a systemic disorder mainly affecting the brain and liver. The lenticular nucleus is most severely affected in the brain, and the name “hepatolenticular degeneration” reflects this pathological finding.1 Neurological manifestations are varied, parkinsonism and dystonia being the most common. As striatal injury was classically described in pathological examination and was repeatedly shown on CT2 3 and MRI,4-7 it has been thought to underlie the parkinsonism and dystonia. Furthermore single photon emission computerised tomography (SPECT)8 and positron emission tomography (PET)9 studies showed loss of D2 receptors in the striatum. A report of poor response to levodopa in a case of Wilson’s disease again supports a postsynaptic cause for the neurological manifestations.10
Whereas there are the unequivocal reports of striatal or postsynaptic lesions, there are no pathological studies demonstrating damage in the substantia nigra.1 11-13 However, there have been clinical and imaging studies suggesting nigral damage in Wilson’s disease; the presence of levodopa responsiveness,14 15 MR studies,5-7 a [18F]-6-fluorodopa PET study,16 and a [11C]-(+)-nomifensine PET study.9 Therefore, we examined whether there is evidence of presynaptic nigrostriatal dopaminergic damage in Wilson’s disease using dopamine transporter (DAT) imaging.
DAT is a protein located mainly in dopaminergic nerve terminals.17 Nuclear imaging using ligands for DAT can measure its density. (1r)-2β-Carbomethoxy-3β-(4-iodophenyl)tropane (β-CIT) is a phenyltropane analogue of cocaine. It binds to DAT with high affinity and low non-specific binding, and may be used as a radiopharmaceutical marker for nigrostriatal dopaminergic nerve terminals in the striatum. Pharmacological characterisation of regional binding disclosed high striatal binding in normal monkeys.18 [123I]-β-CIT SPECT is expected to show nigrostriatal dopaminergic damage, and has been useful for imaging in Parkinson’s disease.19
Therefore we performed [123I]-β-CIT SPECT in neurological patients with Wilson’s disease to examine whether there is presynaptic nigrostriatal dopaminergic damage.
Patients and methods
PATIENTS
We studied six patients with Wilson’s disease (four men and two women). Their ages ranged from 14 to 50 (mean 30). The diagnosis of Wilson’s disease was made by appropriate clinical history of neurological impairment, low serum ceruloplasmin concentration, low serum copper concentration, increased urinary copper excretion, and Kayser-Fleischer rings. All the patients had parkinsonism and dystonia as neurological manifestations. In three patients there were clinical and laboratory findings of liver cirrhosis. MRI was obtained in five patients at the time of diagnosis. Table 1 shows a clinical summary including neurological manifestations at the time of the [123I]-β-CIT SPECT with MR findings.
Clinical summary and MRI findings of the Wilson’s disease group
METHODS
Fifteen normal volunteers (six men and nine women) served as controls. Their ages ranged from 20 to 64 (mean 40). None of them had a history of neuropsychiatric disease or signs of neurological impairment. Six patients with Parkinson’s disease (two men and four women) served as disease controls. The ages ranged from 43 to 60 (mean 53). Their parkinsonian features were mild with Hoehn-Yahr stage 1 or 2. Informed consent was obtained from all the participants after explaining about the experimental nature of the study.
[123I]-β-CIT was prepared from the corresponding trimethylstannyl precursor (Research Biomedicals International, Natick, MA, USA) and high radionuclide purity [123I]-NaI (Korea Atomic Energy Research Institute, Seoul, Korea) using the method described by Zea-Ponce et al with minor modifications.20 Radiochemical purity determined by thin layer chromatography was more than 95% and radiochemical yield was 65±10% (n=5). Apyrogenicity was confirmed by the limulus amebocite lysate test (Endosafe, Charleston, NC, USA). Sterility was confirmed by lack of growth in trypticase and fluid thioglycollate at 35°C for two weeks.
Antiparkinsonian medication in patients with Parkinson’s disease was stopped for 48 hours before the study. All the patients with Wilson’s disease were taking penicillamine. Two were taking zinc sulphate. No patients were taking dopaminergic medication for their neurological symptoms. Zinc sulphate was stopped for 48 hours before the study. Penicillamine was continued. Lugol solution (450 mg) was given to the patients and volunteers one day before the study. [123I]-β-CIT (370 MBq, 10 mCi) bolus injection was done over 30 seconds at 6 00 pm. The SPECT studies were performed using a double headed camera (Prism 2000, Picker, USA) equipped with a medium energy collimator. The patient was in a supine position on the table, and the head was positioned to obtain images in a plane parallel to the orbitomeatal line. The first scans were obtained at one and two hours after injection. On the next day, the images at 18, 21, and 24 hours after injection were obtained. Data were acquired with a 20% symmetric window centred at 159 keV, 128×128 matrix (pixel size 4.67×4.67 mm, slice thickness 4.67 mm) and reconstructed with a Butterworth filter (cut off 0.4 cycle/cm, order 7). For attenuation correction, Chang’s method was used (μ 0.1/cm).
Reconstructed images were used to identify the brain anatomy and measure the radioactivity. Striatal and occipital regions of interest (ROIs) were visually positioned in the three contiguous slices of images showing the highest activity at the striatal region. Striatal ROI was of elliptical shape and of 5×7 pixel size (23.4×32.7 mm) to encompass the whole striatum ; and occipital ROI was positioned within each occipital area in a round shape of 6×6 pixels (28×28 mm). Radioactivities of striatal and occipital ROIs (expressed as counts/pixel/min) in the three slices were summed.
In the occipital lobe, the density of DAT is very low.18Specific striatal binding was defined as the striatal binding minus occipital binding. Striatal/occipital ratios were calculated by dividing the striatal binding of each side by mean occipital binding. The specific striatal/occipital binding ratio (S/O ratio) was calculated by the formula:
Results
The results from measurements at 18, 21, and 24 hours after injection were nearly the same. The results at 18 hours are described. Figure 1 shows the 18 hour SPECT images from a normal volunteer, a patient with Parkinson’s disease, and a patient with Wilson’s disease. In normal controls, [123I]-β-CIT binding was very high in striatal areas and low in other areas including occipital areas as shown in figure 1 (a) in this 30 year old woman. This pattern is consistent with the known distribution of DAT in the monkey.18 In the patients with Parkinson’s disease, [123I]-β-CIT binding was visibly decreased in striatal areas, most marked in the caudal part, compared with that of normal volunteers. Figure 1 (b) shows a 48 year old patient who had hemiparkinsonism on the left side for one year. The decrease in [123I]-β-CIT striatal binding was bilateral, but worse contralateral to the symptomatic side. [123I]-β-CIT binding in the patients with Wilson’s disease was also decreased in striatal areas. As shown in this 26 year old patient 1 (fig 1 (c)), striatal binding was decreased bilaterally and more marked in the caudal part, which was similar to that seen in Parkinson’s disease.
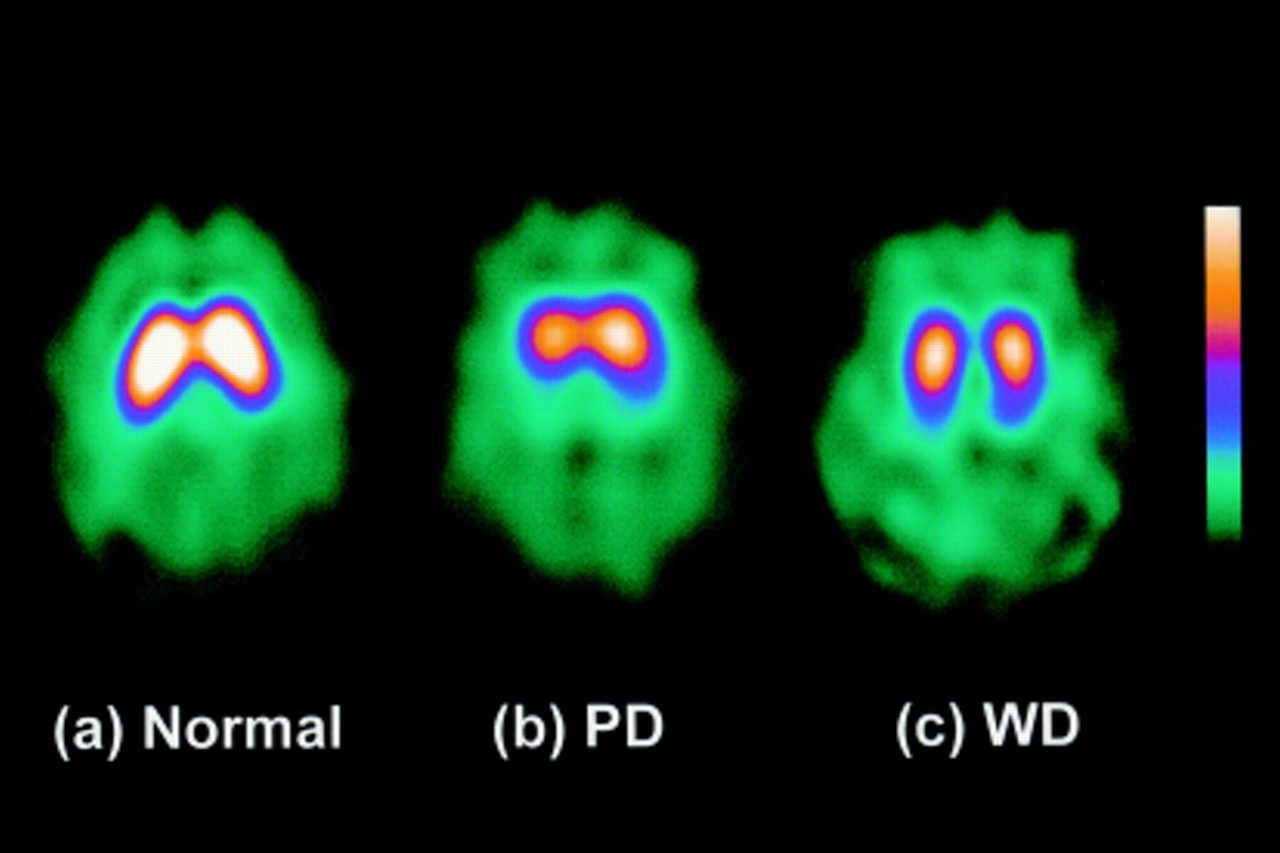
[123I]-β-CIT SPECT images of a normal control (A), a patient with Parkinson’s disease(PD) (B), and a patient with Wilson’s disease (WD) (C). See text for details.
Table 2 shows the specific S/O ratios derived from 18 hour images. In normal controls the specific S/O ratios ranged from 4.52 to 9.67 (mean (SD) 6.22 (1.32)). The ratios in patients with Parkinson’s disease ranged from 2.68 to 4.35 (mean (SD 3.78 (0.65)) and the ratios in the Wilson’s disease group ranged from 2.96 to 4.30 (mean (SD) 3.60 (0.49)). In all patients of the Wilson’s disease and Parkinson’s disease groups, specific binding ratios were below the range of the normal control group without overlap (fig 2). The specific S/O ratios in the Wilson’s disease group were significantly lower than normal control group (p<0.001). Values in the Wilson’s disease group were not different from the Parkinson’s disease group. The specific S/O ratios of normal controls tended to decline with age, as reported previously;21 however, the result was non-significant with the small sample size in this study (r=−0.41, p=0.12). As the Wilson’s disease group was younger than normal controls, the decrease in specific S/O ratio is not explained by age difference.
Summary of the specific striatal/occipital binding ratios in the Wilson’s disease, normal control, and Parkinson’s disease groups

{kind=link}
{kind=link}
Summary of the specific striatal uptake ratios in the normal controls, patients with Wilson’s disease (WD), and patients with Parkinson’s disease (PD). Individual data points are the mean of the right and left striatal/occipital ratios. The ratios in patients with Wilson’s disease and patients with Parkinson’s disease were lower than those in normal controls without overlap. The ratios in Wilson’s disease were essentially in the same range as those in Parkinson’s disease.
Discussion
[123I]-β-CIT binding was high in the striatum in normal volunteers, which is consistent with the information that DAT is high in the striatum. Striatal binding of [123I]-β-CIT was very low in Parkinson’s disease, consistent with damage to the nigrostriatal dopaminergic system in Parkinson’s disease. The symptoms of our patients with Parkinson’s disease were mild and they all were in Hoehn-Yahr stage 1 or 2. However, the density of DAT was significantly decreased bilaterally as previously reported.22 These results indicate that [123I]-β-CIT is a very useful and sensitive probe for nigrostriatal dopaminergic integrity.19 In Wilson’s disease the [123I]-β-CIT binding was far lower than that of normal controls. Decline in DAT with aging21 as seen in our study does not explain the decrease in DAT in patients withWilson’s disease as they were younger than normal controls (mean age 30 in Wilson’s disease, 40 in normal controls). When age matched, the specific S/O ratios in Wilson’s disease were even lower than those of normal controls. Our data show that there is significant nigrostriatal dopaminergic damage in Wilson’s disease. The severity of damage was comparable with that of patients with Parkinson’s disease.
Postsynaptic damage in the striatum has been repeatedly shown in pathological examination,1 11-13 CT or MR images,2-7 and D2 receptor imaging.8 9 Even though striatal injury has been well demonstrated in these studies, clinical correlation between neurological deficits and imaging studies was poor, suggesting that more than striatal lesions play a part in neurological deficits.
There is some evidence suggesting significant presynaptic nigral damage in neurological Wilson’s disease. If parkinsonism in Wilson’s disease is exclusively due to postsynaptic striatal injury, levodopa responsiveness would not be anticipated. But there are some reports of favourable responses to levodopa in the neurological symptoms of Wilson’s disease.14 15
MRI has shown that the distribution of structural changes in the brain is not limited to the striatum.4-7 High signal intensity lesions were found in the midbrain, including the nigral region,5-7 and reversed with penicillamine.7The midbrain signal changes may suggest presynaptic lesions in dopaminergic nerves. In our patients with Wilson’s disease with available MR images, all five patients had signal changes in the midbrain.
In a [18F]-6-fluorodopa PET study by Snowet al,16 the fluorodopa uptake of four neurological patients with Wilson’s disease was decreased in the striatum. Using a DAT ligand nomifensine Westermarket al 9 found reduced striatal binding of [11C]-(+)-nomifensine in three neurological patients with Wilson’s disease. These PET data suggest that significant presynaptic lesions exist.
[123I]-β-CIT SPECT study directly visualises the nigrostriatal dopaminergic nerve terminals and our study shows substantial disruption of dopaminergic nerve terminal integrity in neurological patients with Wilson’s disease. As signal changes were seen in the striatum on MRI, it is almost certain that our patients also have a certain degree of postsynaptic damage. However, we did not examine postsynaptic D2 receptors by [123I]-IBZM SPECT or [11C]-(+)-raclopride PET study.
In conclusion, we showed a decrease of the DAT in the striatum in Wilson’s disease indicating damage to nigrostriatal dopaminergic nerve terminals. There is an apparent incongruity between our data showing that DAT is severely decreased to the Parkinson’s disease range in the striatum and pathological studies showing that there is no damage in the substantia nigra.1 11-13 There may be two explanations for this incongruity. It may be that nigrostriatal dopaminergic damage in Wilson’s disease occurs mainly in the nerve terminals but not in the cell body. It is interesting to note that the dopamine concentration was severely decreased in the striatum when there was no notable pathological change in the substantia nigra in an eight year old patient with Wilson’s disease.13 In another biochemical study, tyrosine hydroxylase activity was decreased in the striatum but not in the substantia nigra.23 These biochemical data are consistent with our data which suggest a preferential damage in the nerve terminals in Wilson’s disease. Another possibility is that DAT is down regulated. Haloperidol24 and reserpine25 decrease the striatal DAT, whereas cocaine26 and mazindol24 increase it. These data suggest that the DAT is subject to dopaminergic regulation, even though levodopa itself does not change the DAT.24 Penicillamine and zinc sulphate were the only medications in the patients with Wilson’s disease. Penicillamine and zinc sulphate do not have known dopaminergic action, thus are unlikely to regulate the DAT directly. However, the absence of direct information on penicillamine, zinc sulphate, and DAT makes it difficult to completely rule out this possibility. It is known that DAT gene expression is increased by direct interaction with target striatal cells in culture of rat embryonic mesencephalic dopaminergic neurons.27 Striatal injury in Wilson’s disease may cause a loss of synaptic contact for nigrostriatal dopaminergic nerve terminals resulting in down regulation of the DAT and tyrosine hydroxylase. However, hypoxic-ischaemic striatal injury in the neonatal rat does not decrease striatal dopamine,28 dopamine uptake sites measured by [3H]mazindol binding,28 TH immunostaining,29 and in vivo tyrosine hydroxylase activity.30 Nor does axon sparing striatal excitotoxic injury in the adult rat result in decreased tyrosine hydroxylase activity31 and tyrosine hydroxylase immunostaining.32 We interpret these data to mean that loss of postsynatic neurons in the striatum does not result in down regulation of the DAT and tyrosine hydroxylase at least after birth. Therefore, we favour the hypothesis that decrease of DAT in Wilson’s disease is due to direct nerve terminal damage, not due to down regulation from postsynaptic striatal injury.
The degree of presynaptic damage measured by in vivo DAT imaging was comparable with that of Parkinson’s disease, and may well have a profound influence on the neurological manifestations of Wilson’s disease as well as postsynaptic lesions. The relative proportion of presynaptic and postsynaptic damage may explain conflicting reports of levodopa responsiveness.
Acknowledgments
This study was in part supported by the Korea Science Foundation (95–0403–96–3). We express thanks to participants in the study including the chief residents in the Department of Neurology, and to Dr Paul Greene at Columbia-Presbyterian Medical Center in New York for reading the manuscript and giving thoughtful suggestions.