
Abstract
BACKGROUND AND PURPOSE: Sarcoidosis is an idiopathic systemic granulomatous disease, recognized in a patient when clinical and radiologic findings are confirmed by histopathologic analysis. The objective was to identify a relationship between MR imaging and clinical findings in CNS sarcoidosis.
METHODS: The clinical charts of 461 patients with biopsy-proved sarcoidosis were reviewed retrospectively. Criteria for including patients in the study included those with symptoms referable to the CNS, excluding those with another explanation for their symptoms, those with headaches or other subjective complaints without accompanying objective findings, and those with peripheral neuropathy other than cranial nerve involvement or myopathy without CNS manifestations. Thirty-four of 38 patients whose conditions met the criteria for CNS sarcoidosis underwent a total of 82 MR examinations. The positive imaging findings were divided into categories as follows: pachymeningeal, leptomeningeal, nonenhancing brain parenchymal, enhancing brain parenchymal, cranial nerve, and spinal cord and nerve root involvement. Treatment response, clinical symptomatology, and any available histopathologic studies were analyzed with respect to imaging manifestations in each of the categories.
RESULTS: Eighty-two percent of the patients with sarcoidosis with neurologic symptoms referable to the CNS had findings revealed by MR imaging. However, eight (40%) of 20 cranial nerve deficits seen at clinical examination of 13 patients were not seen at contrast-enhanced MR imaging, and 50% of the patients with symptoms referable to the pituitary axis had no abnormal findings on routine contrast-enhanced MR images. In contradistinction, 44% of 18 cranial nerves in nine patients with MR evidence of involvement had no symptoms referable to the involved cranial nerve. Clinical and radiologic deterioration occurred more commonly with leptomeningeal and enhancing brain parenchymal lesions.
CONCLUSION: MR imaging can be used to confirm clinical suspicion and to show subclinical disease and the response of pathologic lesions to treatment.
Sarcoidosis is an idiopathic systemic granulomatous disease, recognized in a patient when clinical and radiologic findings are confirmed by histopathologic evidence of noncaseating granulomas (1). Clinical neurologic involvement occurs in approximately 5% of patients. However, autopsy series that have shown neurologic involvement in 14% to 27% of patients with sarcoidosis suggest that subclinical involvement may be present in substantially more (2–4).
Neurosarcoidosis has a variable expression, which tends to mimic other neurologic diseases. The diagnosis is based on the documentation of systemic sarcoidosis in the absence of other neurologic disease (4). The histopathologic hallmarks include epithelioid granulomas without caseation or staining for infectious agents. These granulomas often incorporate multinucleated giant cells and lymphocytes (5–7). Compared with sarcoidosis granulomas elsewhere in the body, these tend to be smaller, have fewer multinucleated giant cells, and be associated with blood vessels (8, 9). The frequent simultaneous expression of new and old granulomas suggests that the disease process waxes and wanes (2).
Methods
We retrospectively reviewed the clinical charts of 461 patients who had biopsy-proved sarcoidosis and who underwent treatment for sarcoidosis at the Henry Ford Hospital between 1985 and 1995. We identified those patients with CNS manifestations. Exclusion criteria included other known neurologic causes for the symptoms, headache or other subjective complaints without accompanying objective findings, and peripheral neuropathy (other than cranial nerve involvement) or myopathy without CNS manifestations. Patients with neurologic symptoms were examined by the appropriate specialist (ie, neurosurgeon, neurologist, neuroophthalmologist, endocrinologist, etc) depending on the symptoms.
Thirty-four patients underwent a total of 82 MR examinations, 81 of which included contrast-enhanced sequences. All MR examinations were performed at a field strength of 1.5 T. Each patient underwent at least one examination after contrast administration. Imaging sequences of the brain (60 studies) included long TR spin-echo sequences, short TR spin-echo sequences, and contrast-enhanced short TR sequences in multiple planes. Spinal MR examinations (18 studies) included sagittal and axial spin-echo short TR sequences, fast spin-echo T2-weighted or spin-echo T2-weighted sequences, and contrast-enhanced T1-weighted spin-echo sequences.
The patients were divided into six groups according to imaging manifestations, which included dural meningeal involvement, leptomeningeal involvement, nonenhancing brain parenchymal lesions, enhancing brain parenchymal lesions, cranial nerve involvement, and spinal involvement. Within each category, the clinical symptoms and response to treatment were analyzed and compared.
Patients were considered to have dura-based sarcoidosis intracranially when imaging showed the dura-arachnoid pattern as described by Meltzer et al (10). Within the spine, an extraaxial intradural lesion attached to the thecal sac was also considered to be a dura-based lesion. Because of the difficulties in distinguishing leptomeningeal cord involvement from intrinsic cord involvement, only leptomeningeal lesions involving the brain were included under the category of leptomeningeal involvement. Leptomeningeal lesions were considered as such if the pia-subarachnoid pattern described by Meltzer et al could be identified. A patient was considered to have an enhancing brain parenchymal lesion if intraparenchymal enhancement was clearly demonstrated. A patient was considered to have a nonenhancing brain lesion if such a lesion was clearly evident and was entirely separate from any areas of contrast enhancement. Cranial nerve lesions were considered to be present when acquired abnormalities of the cranial nerves without evidence of cause other than sarcoidosis were observed. Spinal lesions were considered to be present when intrathecal spinal involvement was noted. The results of the clinical and MR examinations performed after treatment were reviewed, when available, and compared.
Results
The 461 patients with biopsy-proved sarcoidosis included 288 (62%) women and 173 (38%) men. Three hundred one (65%) were African American, 151 (33%) were white, three (0.7%) were Indian, two were Arabic (0.4%), and two were Hispanic (0.4%). A total of 38 (8.3%) patients fit the stringent criteria for CNS sarcoidosis as described herein. They included 20 (53%) men and 18 (47%) women. Twenty-nine (76%) were African American, seven (18%) were white, one (2.6%) was Arabic, and one (2.6%) was Hispanic. The average age at the time of presentation of sarcoidosis was 33.7 years (standard deviation [SD], 6.1 years). The average patient age at the time of presentation with neurologic symptoms was 35.3 (SD, 6.9 years).
Of these 38 patients, 24 (63%) had neurologic symptoms as their first manifestation of sarcoidosis. The other 14 patients (37%) had neurologic symptoms an average of 56 months (SD, 50 months) after the initial diagnosis of systemic sarcoidosis. Four (11%) of 38 patients had sarcoidosis confined to the nervous system. Systemic involvement included pulmonary in 31 (82%) of the 38 patients, skin in 11 (29%), uveitis in seven (18%), liver in four (11%), spleen in two (5.6%), parotid in two (5.6%), and cardiac in two (5.6%). Other systemic involvement included lacrimal, stomach, larynx, and nasal septum. The measurements of serum angiotensin-converting enzyme were elevated in nine (50%) of the 18 patients in whom it was measured. In seven of nine patients in whom CSF was examined, protein and WBC were elevated. Eight patients underwent a total of 10 biopsies of CNS lesions.
Thirty-four patients with neurologic symptoms underwent MR imaging. Nineteen (56%) of the patients were male and 15 (44%) were female. Twenty-five (74%) were African American, seven (21%) were white, one (3%) was Arabic, and one (3%) was Hispanic. Twenty-eight (82%) of 34 patients had positive MR findings. These 28 patients were divided into six categories based on imaging findings (as described herein), and the results were analyzed within each category. These results are presented in Tables 1 through 6⇓⇓⇓⇓⇓. The clinical and MR follow-up data obtained after treatment are summarized in Tables 7 and 8.
Dural meningeal sarcoid involvement
Leptomeningeal sarcoidosis involvement
Enhancing brain parenchymal lesions
Nonenhancing brain parenchymal lesions
MRI involvement of cranial nerves
Spinal cord and nerve root sarcoid involvement
Clinical follow-up of CNS sarcoidosis
MR follow-up of CNS sarcoidosis
Clinical follow-up was conducted for 26 patients for an average of 60 months (range, 7 to 176 months). Seven (27%) of 26 patients experienced complete resolution of their symptoms after treatment, without any clinical recurrence. Five (19%) of 26 patients experienced improvement, three (12%) had no change, and eight (31%) had recurrences of either similar or unrelated symptoms of CNS sarcoidosis after first showing resolution of their symptoms; two of the eight patients went on to complete resolution of their symptoms. Two (8%) of 26 patients experienced a deterioration of their conditions without any period of remission.
MR follow-up was conducted for 19 patients for an average of 27 months (range, 2 to 110 months). No patients had complete resolution of their MR findings without any evidence of recurrence. Five (26%) of 19 had regression of their MR imaging abnormalities. Six (32%) of 19 had no change. Four (21%) of 19 had evidence of recurrence after improvement; one (25%) of the four patients went on to complete resolution of the MR findings. Four (21%) of 19 patients experienced further progression of their disease, as revealed by MR imaging.
Eight patients with dural involvement presented either with headache (n = 4, 50%) or with symptoms of nerve compression (n = 5, 62%). Clinical resolution was more frequent than in the other categories in our study, and dural recurrence was not experienced. Four of these eight patients underwent biopsy of the lesions, which revealed epithelioid granulomas within a bed of fibrocollagenous material. These lesions were negative for acid-fast bacillus (AFB) and fungal stains. The fibrocollagenous material was suspected, at least in part, to be responsible for the lower signal intensity on T2-weighted images (Fig 1).

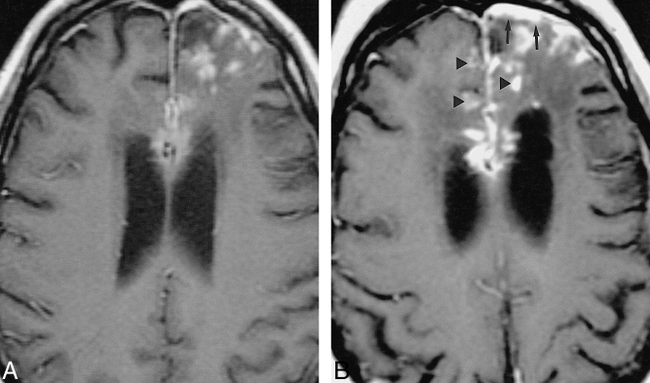
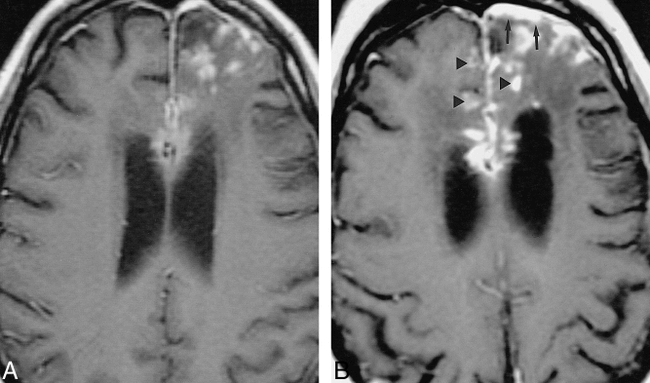
33-year-old white woman with chronic headache.
A–D, An extraaxial left parietal mass is hypointense on noncontrast T2-weighted MR image (2517/90/1 [TR/TE/excitations]) (A), enhances on contrast-enhanced T1-weighted (533/11/2) image (arrows, B), and is isointense with gray matter on noncontrast T1-weighted image (450/19/2) (arrows, C). Substantial fibrocollagenous material and noncaseating granulomas were seen at biopsy. The patient's symptoms resolved with steroids; however, a T1-weighted image (400/16/2) 28 months later showed only partial regression of the lesion (D).
Patients with leptomeningeal lesions presented with a variety of symptoms that could be accounted for, at least in part, by the location of the MR findings and the involvement of adjacent structures (Table 2). Although three (60%) of five patients (Tables 2 and 7) experienced at least partial if not complete resolution of their symptoms after steroid treatment, all three patients had clinical recurrence with interim periods of reprieve of 9, 13, and 72 months, respectively. Imaging revealed a clear predilection for suprasellar basal (n = 4, 80%) and, to a lesser degree, frontal basal (n = 2, 40%) leptomeninges (Fig 2). MR findings and follow-up data correlated well with clinical symptoms and clinical response. Two patients with leptomeningeal involvement underwent biopsy, which showed epithelioid granulomas infiltrating the leptomeninges and adventitial layers of the blood vessels within, with negative staining for microorganisms.

33-year-old African-American woman with right-sided visual loss, panhypopituitarism, polydipsia, and polyuria (with normal ADH).
A and B, T1-weighted contrast-enhanced MR image (400/15/2) (A) shows basal meningeal enhancement and an enhancing pituitary mass involving both lobes, the infundibulum, and the right optic nerve (not shown). Pituitary and meningeal biopsy revealed noncaseating granulomas with negative AFB and fungal stains. The visual symptoms resolved with steroid treatment; however, 21 months later she returned with a left abducens palsy. A T1-weighted image (400/12/2) shows resolution of the pituitary lesion but new involvement of the left cavernous sinus (arrows, B).
The most common clinical symptom in the patients with enhancing brain lesions was seizures (six of eight patients, 75%) (Table 3). Although a certain degree of generalized volume loss is expected with steroid use, delayed focal atrophy was noted to occur in two patients with persistent brain parenchymal lesions within 95 and 172 months, respectively (Fig 3). Neither of these patients received effective steroid treatment. One of the two developed diabetes type II, which limited steroid dose, and the other patient's diagnosis of CNS sarcoidosis was delayed 172 months. Good MR and clinical correlation was established; however, MR improvement lagged behind clinical improvement on follow-up examinations. Parenchymal biopsy specimens obtained from one patient on two separate occasions showed noncaseating granulomas, lymphocytes, and histiocytes involving the perivascular spaces, and adventitial layers of the vessels but not occluding most vessels (Fig 4). Although lymphocytic infiltration into the adjacent brain parenchyma was present, reactive astrogliosis and microglial unrest was also evident. Special stains failed to demonstrate AFB, fungi, spirochetes, or other bacteria. No large brain infarcts were seen in any of our patients. Of note, four (50%) of eight patients with brain parenchymal lesions had uveitis. Among our 38-patient CNS sarcoidosis population, seven (18%) had uveitis.
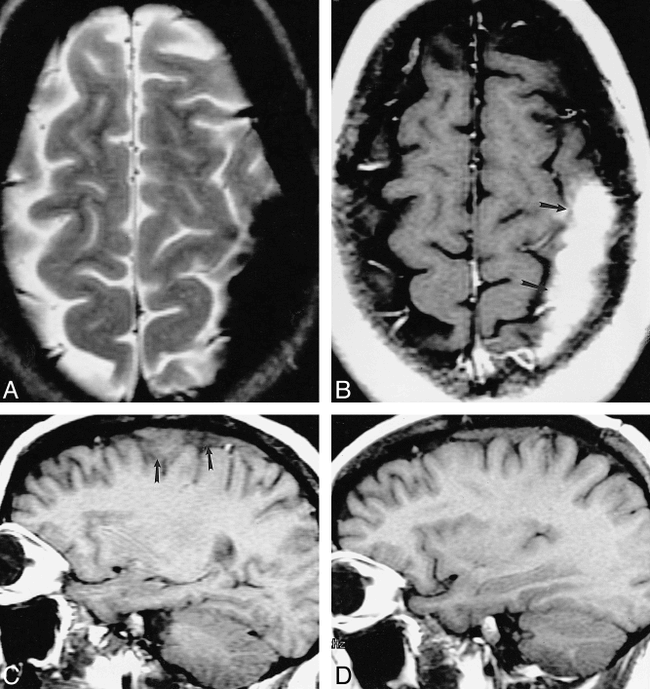
52-year-old African-American man with seizure disorder. Imaging revealed hydrocephalus and a brain parenchymal lesion. His seizures gradually became more difficult to control.
A and B, Contrast-enhanced T1-weighted MR images obtained 9 (483/13/2) (A) and 11 (500/16/2) (B) years after seizure onset show interim progression of meningeal thickening (arrows, B) and enlargement and multiplication of enhancing foci (arrowheads, B).

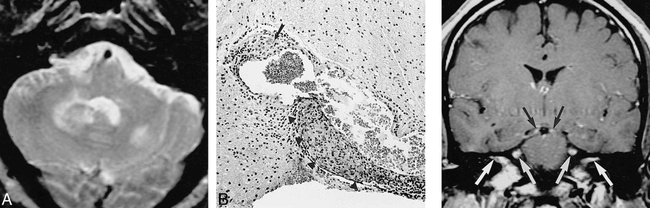
34-year-old Arabic man with seizures, dysphagia, and lower extremity weakness.
A, T2-weighted MR image (2416/90/1) shows multiple hyperintense lesions in the brain stem and cerebellum as well as the cerebral hemispheres and basal ganglia (not shown), which demonstrated contrast enhancement.
B, Biopsy specimen shows atypical lymphoid tissue with noncaseating granulomatous plaques (arrow) and granulomatous perivasculitis (arrowheads) (hematoxylin-eosin). The symptoms and MR abnormalities disappeared 15 months after treatment with steroids and hydroxychloroquine sulfate.fig 5. 36-year-old African-American man with lower extremity weakness, hypopituitarism, mild unilateral trigeminal dysesthesia, and anosmia. Contrast-enhanced T1-weighted MR image (700/19/2) shows enhancement of trigeminal oculomotor nerves and within both internal auditory canals. The patient's symptoms resolved with steroids but recurred upon tapering of the medication. The symptoms were never referable to any of the MR findings.
Patients with nonenhancing brain parenchymal lesions presented with a variety of symptoms, which in five of six cases were thought not to correlate with the clinical symptoms (Table 4). The appearance of these lesions on the images tended to mimic demyelinating disorders. CSF study revealed positive oligoclonal bands for one patient. In general, these patients achieved better clinical outcomes than did the patients with enhancing brain parenchymal lesions.
A total of 13 patients manifested cranial nerve symptoms, and nine patients had MR findings referable to cranial nerves. Of 20 clinically detected cranial nerve deficits in 13 patients, only eight (40%) had correlative MR findings. On the other hand, of 15 enhancing cranial nerves and three enhancing optic chiasms, only eight (44%) had corresponding cranial nerve deficits (Table 5). Of four patients with good treatment response and follow-up MR imaging, three (75%) had persistent MR findings. These results indicate that cranial nerve involvement evidenced by imaging does not always imply a clinical deficit (Fig 5). The most common cranial nerve deficit was that of cranial nerve VII (seven of 13 patients, 54%); the next most common were those of cranial nerve II (n = 13, 38%) and cranial nerve V (n = 3, 23%). Other clinical deficits included those of cranial nerve I (one patient), cranial nerve III (two patients), cranial nerve VI (two patients), cranial nerve X (one patient), and cranial nerve XII (one patient). On MR images, the most common abnormality involved cranial nerve II (five of nine patients, 55%), considering chiasm as part of cranial nerve II. Excluding cranial nerve II, all five cranial nerve deficits, with corresponding MR findings of cranial nerve enhancement, showed a clinical response to treatment. Four patients with cranial nerve findings underwent biopsy. A representative case showed granulomas with epithelioid histiocytes and associated lymphoid infiltrate but without necrosis. A neurofilament antiserum revealed axons near the granuloma, and GFAP stain disclosed prominent astrocytosis. AFB, bacterial, and fungal stains were negative.
Patients with spinal sarcoidosis usually manifested symptoms of weakness (six of seven patients, 86%) (Table 6). Patients with both nerve root and cord involvement were included. Of four cord lesions, three manifested enhancement, predominantly along the periphery of the cord but also extending toward the center of the cord (Fig 6), and significantly more cord signal abnormality on the T2-weighted images. The fourth patient had a focal cord signal abnormality in the right hemicord, with minimal enhancement along the periphery. Only this latter patient had complete resolution of her symptoms; the other patients experienced only partial improvement. Patients with nerve root involvement had a dura-based lesion compressing the nerve and/or nerve root enhancement. Of the three patients with these symptoms, one had complete resolution and two had partial or no improvement. MR follow-up was limited to cord lesions in this group, which, in both cases, showed a waxing and waning process in both symptoms and MR findings in which MR improvement lagged behind symptomatic improvement. Focal cord atrophy eventually developed in both cases. Biopsy results in a patient with cord involvement were similar to those of enhancing brain parenchymal lesions in which both granuloma formation and lymphocytic infiltrate were identified in the leptomeninges, perivascular spaces, and adjacent cord with associated glial reactive process and adventitial layer vascular infiltration. Biopsy results in a patient with a dura-based lesion involving a nerve root, as revealed by noncontrast MR imaging, showed noncaseating granulomatous involvement in the epidural space, arachnoid, and cauda equina.

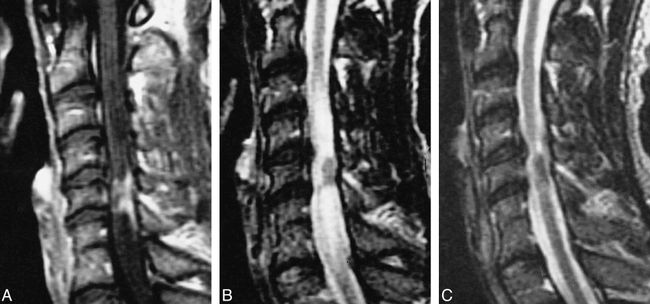
36-year-old African-American man with gradual onset of weakness, numbness, spasticity, and bowel and bladder dysfunction.
A–C, T1-weighted MR image (450/15/2) (A) shows intramedullary enhancement and enhancement along the surface of the cord, associated with cord signal abnormality and disk herniation on T2-weighted fast spin-echo image (B). The patient underwent surgical diskectomy and initially did worse. His symptoms and the findings on a T2-weighted fast spin-echo image (3000/102/2) (C) improved with steroids, but recurred 5 months later.
Seven patients with clinical but not imaging findings were identified in this study. Four (57%) had cranial nerve deficits, one had diabetes insipidus, one had hypopituitarism, encephalopathy, and headache, and one had myelopathy. Because of the small size of the structures involved and the normal enhancement of the pituitary, it is possible that the lesions were below the threshold of imaging.
Discussion
The pathophysiological mechanism behind sarcoidosis remains elusive. In theory, it is thought to represent an immune-mediated response to an as yet unidentified antigen. Both an acute self-limited coarse with spontaneous resolution and an insidious, relentless course ultimately resulting in fibrosis have been identified. The disease most commonly afflicts African Americans (female > male) in the third to fourth decade of life but can occur in every race and has a wide age range. Previous imaging experiences are summarized in Table 9 (11, 12).
MR manifestations of CNS sarcoidosis
What we offer in this study is a description of 38 cases of CNS sarcoidosis derived from a 461-patient population. Epidemiological data were reviewed, and patients were divided into six categories on the basis of imaging location. MR and clinical findings were reviewed to determine what MR findings mean clinically. In comparing clinical and MR improvement, the retrospective approach may bias toward imaging patients with more serious symptoms or patients whose symptoms have not resolved. We do demonstrate, however, that there are clear differences between clinical symptoms and imaging findings. In general, the radiologic presence of CNS sarcoidosis is not always evident clinically. Whereas 46% of patients improved or had complete clinical resolution of their symptoms, only 26% of patients did so on the basis of imaging. This is supported by the higher prevalence of CNS sarcoidosis identified in autopsy series than in clinical series (2–4).
Dural Thickening/Mass
Dural mass lesions had an imaging appearance similar to that of meningioma and were not associated with intraparenchymal extension. The annual incidence of these lesions in our 461-patient population exceeds the annual incidence of meningiomas (2.3 per 100,000) (13) by 130 times. These lesions tend to be isointense with gray matter on T1-weighted MR images and hypointense on T2-weighted images, and they enhance uniformly. This hypointensity on T2-weighted images has been reported previously (14, 15) and is thought to be related to fibrocollagenous buildup. This is supported by histopathology of lesions biopsied within our series. Unfortunately, this is not a unique finding. Eighteen percent of meningiomas (typically fibroblastic or transitional types) demonstrate low signal on T2-weighted images (16). Patients with dura-based lesions in our study typically presented with headaches or symptoms of nerve compression and generally responded symptomatically to treatment. Imaging revealed that treatment had partially but not completely diminished the size of lesions observed. In addition to meningioma, differential considerations for sarcoidosis involvement of the dura should include other causes of chronic meningitis, such as lymphoma, adenocarcinoma, Wegener's, idiopathic hypertrophic cranial pachymeningitis (IHCP), granulomatous infection, and leukemia. Cheng et al (17) reviewed the cases of 37 patients who underwent biopsy for chronic meningitis. Of the 16 diagnostic biopsies in this study, 31% revealed sarcoidosis and 25% revealed adenocarcinoma. Although most patients with neurosarcoidosis have systemic involvement (2), isolated dural involvement may be confused pathologically with IHCP because both are granulomatous diseases negative for AFB or fungal staining. Unlike IHCP, which tends to manifest more diffuse involvement, dural sarcoidosis is often focal and has nonnecrotizing epithelioid granulomas (18–20).
Leptomeningeal Involvement
The relatively low rate of occurrence of combined dural and leptomeningeal disease in the same location can be explained by the presence of the arachnoid barrier cells. This portion of the arachnoid mater lacks extracellular spaces, has numerous cell junctions, and has a basement membrane separating it from the subarachnoid space. It can thus act to slow down or prevent the infiltration of lymphocytic cells (10, 21). In this series of 12 patients with either dural or leptomeningeal involvement, only one (8%) had involvement of both. In this case, the patient first presented with pituitary and right optic nerve involvement and, 18 months after treatment and resolution of symptoms, presented with a right cranial nerve VI palsy. It can be argued, however, that the disease spread along the carotid artery and the inferolateral trunk and not directly through the dura. Finelli et al (18) reported an unusual case of sarcoidosis involvement of the calvaria, meninges, and underlying brain; however, this should be regarded as the exception rather than the rule. Leptomeningeal sarcoidosis, as we found in this study, is often associated with enhancing brain parenchyma lesions (four of five, 80%) and cranial nerves (three of five, 60%). Biopsy has established an intimate relationship to the vascular elements of this layer as well as the perivascular space, which can thus act as a conduit for disease infiltration (5, 22).
Leptomeningeal involvement is best shown on spin-echo contrast-enhanced T1-weighted images (23). Leptomeningeal infiltration typically involves the suprasellar and frontal basal meninges but may occur anywhere and is more concentrated in the depths of the sulci (5). Granulomatous lesions are accompanied by varying degrees of fibrosis and hyalinization (6). Occasionally, granulomas coalesce to form masslike lesions, particularly in the region of the chiasm, floor of the third ventricle, and pituitary stalk (2). Clinical symptoms correlate with the location of the lesion on MR images. Furthermore, we found that these lesions recur frequently. Disease entities that can involve the basal leptomeninges and mimic leptomeningeal sarcoidosis on imaging include granulomatous disease (such as tuberculosis), Wegener's granulomatosis and fungal meningitis, pyogenic meningitis, leptomeningeal lymphoma, demyelination, meningoangiomatosis, acute lymphocytic leukemia, and leptomeningeal carcinomatosis (10).
Enhancing Brain Parenchymal Lesions
The frequent association of intraparenchymal granulomas with small arteries and veins suggests that lesions infiltrate the brain through perivascular spaces (6, 24, 25). This usually occurs along the adventitia, but the infiltration may, on occasion, destroy the elastic lamina and encroach on and occlude the vessel lumen, potentially causing infarction (5). It is often difficult to distinguish sarcoidosis angiitis from primary isolated angiitis of the CNS on a histopathologic basis (6, 26, 27). Anecdotally, in our series, patients with enhancing brain parenchymal lesions had a higher frequency of uveitis. The meaning or validity of this finding without a larger patient population is uncertain.
Although no part of the CNS is immune to sarcoidosis, frequently affected brain parenchymal locations include the hypothalamus, brain stem, cerebral hemispheres, and cerebellar hemispheres (5, 6, 24–26, 28). Granulomatous infiltration into the subependymal layers of the ventricular system is thought to be responsible for hydrocephalus associated with neurosarcoidosis (5, 6, 12, 25). Involvement of the pituitary is less common.
The most common symptom occurring with enhancing brain parenchymal lesions is seizures, although headache, encephalopathy, diabetes insipidus, and hypopituitarism also occur. Seizures have previously been associated with poorer prognosis in neurosarcoidosis (29), and this has been supported by our results. Patients with enhancing brain lesions had recurrence of symptoms, worsening of symptoms, or no change in 75% of the cases. Histopathologically, this disease infiltrates via the perivascular spaces to reach the brain parenchyma. It would thus be reasonable to hypothesize that if the disease is revealed by imaging to predominantly involve leptomeninges with little parenchymal involvement, it is at an earlier stage. The clinical course in patients with either leptomeningeal or parenchymal disease was hampered by recurrences or deterioration. Although diabetes insipidus is often identified in neurosarcoidosis, it is frequently associated with a normal vasopressin level, thus implying hypothalamic dysfunction (30).
Nonenhancing Brain Parenchymal Lesions
These lesions tend to occur in the periventricular white matter but may also occur in the brain stem and basal ganglia. Periventricular lesions are nonspecific and can occur in association with multiple sclerosis, hypertension, and vasculitis. Theoretically, they can result from periventricular granulomas or from small areas of infarction caused by granulomatous angiopathy. The exact nature of these lesions has not been identified in the literature. They are not as often associated with leptomeningeal or enhancing brain parenchymal lesions, and we suggest that they could differ pathologically from enhancing lesions that infiltrate basal meninges and associated blood vessels, thus disturbing the blood-brain barrier. Of interest is that elevated oligoclonal bands in the CSF, typically associated with multiple sclerosis, are often seen with neurosarcoidosis as well (31). In our patient group, symptoms associated with these lesions did not correlate well with imaging findings. Symptomatic improvement usually did not correspond to improvement on MR imaging studies. This behavior differed from that of enhancing brain lesions for which symptoms often correlated with imaging findings and symptomatic improvement correlated with regression on MR images. These facts also imply that the nonenhancing brain parenchymal lesions and the enhancing lesions have different pathophysiologic mechanisms.
Cranial Nerve Involvement
Involvement of every cranial nerve has been described in association with sarcoidosis (2). Most frequently, the facial nerve is involved clinically. The annual incidence of Bell's palsy in the general population is 25 per 100,000 (32). Among our 461 patients with sarcoidosis, it was 14 times that. Imaging, however, revealed that the optic nerve and/or chiasm are the most frequently affected cranial nerves.
Clinical and imaging cranial nerve involvement frequently do not concur, and, furthermore, clinical resolution often does not imply imaging resolution. Although most patients showed response to steroids, patients with optic nerve involvement often had residual symptoms or no response to treatment. We conjecture that this may be related to the fact that other cranial nerves are surrounded by Schwann cells, which can regenerate more readily than can oligodendroglial myelin, thus allowing for more effective regeneration of function (9).
Cranial nerve involvement is not well understood. Pathologic examination has revealed perivascular and intraneural lymphocytic infiltration (33), as in our results. Nerve root and cranial nerve involvement has been shown by others, as well as by our study, to be caused by compressive effects from adjacent granulomas (4).
Spinal Cord and Nerve Root Involvement
Spinal cord and nerve root involvement has been reported as an unusual manifestation of sarcoidosis. However, we found such involvement in eight of 34 patients who underwent imaging. Furthermore, the majority (seven of eight) of the patients were male. This is especially interesting because sarcoidosis more frequently affects females. Most patients in our series improved with steroid or cyclophosphamide (Cytoxan) administration; however, long-term steroid dependence with episodes of recurrence associated with tapering the administration occurred in three of four patients with cord lesions and in one of three patients with nerve root involvement. Improvement on MR images lagged behind steroid treatment. In concurrence with our results, a review of the literature by Nagai et al (34) found that early treatment with steroids can result in remarkable recovery; however, with a delayed diagnosis and treatment, the disease typically only partially resolves and may recur. Nagai et al additionally commented that decompressive laminectomy as well as lysis of adhesions may also benefit the patient.
Histopathologic specimens have shown granuloma formation with lymphocytic infiltrates. Lesions usually have an extramedullary component; however, pure intramedullary tumors have been described (35). The lesions have been found to represent granulomatous meningitis with nodular studding infiltrating the perivascular space and forming intramedullary granulomas (34). Lesions have been reported to occur throughout the spinal cord but more frequently in the cervical spine (34, 36). At imaging, they manifest cord swelling, with increased signal intensity on long TR images and a pattern of enhancement that predominates in the periphery of the cord but includes patchy multifocal enhancement of the cord. Imaging findings are thus nonspecific and can mimic multiple sclerosis, cord tumor, vacuolar myelopathy, tuberculosis, or fungal infection (36–39). Junger et al (38) hypothesized that patients with spinal cord sarcoidosis progress in four phases, which begin with a linear leptomeningeal pattern of enhancement and progress to a phase in which there is cord enlargement with faint enhancement or no enhancement. Enhancement then progresses but the cord begins to reduce in size until it reaches a final stage of atrophy without any enhancement. Although it is fairly evident that patients who present with cord atrophy will most likely not respond to steroid treatment, it is unclear whether the degree of enhancement plays a role in treatment response. Of our eight patients with spinal involvement, five had concurrent intracranial imaging; one of the five had clinical and imaging findings referable to the cranial nerves (see Table 6). Another patient was found to have brain parenchymal disease 8 years later. Concurrent intracranial disease in patients with spinal sarcoidosis thus does not seem to be significantly different from that of other patients with sarcoidosis.
Conclusion
CNS sarcoidosis is a disease that can involve any part of the CNS, producing varied clinical and MR expressions. It can mimic other neurologic diseases, such as multiple sclerosis, tuberculosis, isolated angiitis of the CNS, and meningioma, and is often indistinguishable from them. Patients with symptoms usually have corresponding CNS lesions on MR images. However, patients with cranial nerve deficits and pituitary dysfunction often have no abnormal findings at routine contrast-enhanced MR imaging. On the other hand, MR findings may be clinically silent. Patients with dura-based lesions, cranial nerve lesions, and, to a lesser degree, nonenhancing brain lesions faired better than did patients with leptomeningeal, brain parenchymal, and spinal lesions. In general, resolution of lesions on MR imaging lags behind resolution of clinical symptoms. The role of MR imaging in neurosarcoidosis is to confirm clinical suspicion, establish subclinical disease, and document the response of pathologic lesions to treatment.
Footnotes
References
- Received May 18, 1998.
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Isolated neurosarcoidosis with a primary lesion in the cauda equina
- Spinal Cord Sarcoidosis Occurring at Sites of Spondylotic Stenosis, Mimicking Spondylotic Myelopathy: A Case Series and Review of the Literature
- Vascular Involvement in Neurosarcoidosis: Early Experiences From Intracranial Vessel Wall Imaging
- Clinical and MRI phenotypes of sarcoidosis-associated myelopathy
- The spectrum of immune-mediated and inflammatory lesions of the brainstem: Clues to diagnosis
- Neuroimaging of Rapidly Progressive Dementias, Part 2: Prion, Inflammatory, Neoplastic, and Other Etiologies
- Correlation of MR Imaging Findings and Clinical Manifestations in Neurosarcoidosis
- Relapsing encephalopathy with headache: an unusual presentation of isolated intracranial neurosarcoidosis
- Impact of [18F]-fluorodeoxyglucose ([18F]-FDG) imaging in sarcoidosis: unsuspected neurosarcoidosis discovered by [18F]-FDG PET and early metabolic response to corticosteroid therapy
- Spinal Cord Involvement in Primary Angiitis of the Central Nervous System: A Report of Two Cases