
Abstract
Summary: We report a case of genetically verified Charcot-Marie-Tooth disease in which the patient had cranial nerve symptoms. CT and MR imaging demonstrated enlargement of several cranial nerves, as well as their skull-base foramina, with faint contrast material enhancement identified.
Charcot-Marie-Tooth (CMT) disease is one of the hereditary motor and sensory neuropathy (HMSN) disorders, a group of genetically based disorders characterized by progressive motor weakness, decreased nerve conduction velocities, and nerve root enlargement (1). Although these disorders are most often diagnosed clinically, MR and CT imaging in these patients is being used more frequently. The bulk of the imaging literature of this disorder concerns the identification of nerve root enlargement, often massive, within the lumbosacral spine in patients with nerve compression syndromes (2–5). Involvement of cranial nerves, both clinically and radiographically, is thought to be quite rare. The following case illustrates involvement of several cranial nerves in a patient with CMT disease.
Case Report
At the time of initial presentation to our institution, the patient was a 64-year-old man with a history of lower extremity weakness that began in his second decade. Weakness had progressed over time and was associated with significant muscle wasting of both lower extremities and hands. The patient also had bilateral deafness and severe right facial pain diagnosed as trigeminal neuralgia. The trigeminal neuralgia had been treated in the past with radio-frequency ablation and alcohol injection, but the patient had recurrent and severe symptoms (6).
Of interest, review of the patient’s family history revealed two similarly affected relatives in an autosomal dominant pattern of inheritance (Fig 1). The diagnosis of CMT was verified with use of electrophysiologic testing, as well as genetic analysis (the latter was also performed on a total of 13 persons in the patient’s family). A novel deletion in the PMP22 gene was found in our patient (6).

Limited family tree demonstrates the affected members of the patient’s family, with current patient marked with arrow. (Modified from reference 6).
Both thin-section CT and MR imaging were performed. CT images demonstrated enlargement of multiple skull-base foramina (Fig 2), whereas the MR images better demonstrated enlargement of the cranial nerves themselves (Fig 3).

A–C, Thin-section (1-mm) coronal and axial CT images of the skull base obtained with an edge-enhancing bone algorithm show enlargement (arrows) of the mastoid canals of cranial nerve VII (A), the foramina of ovale (B), and rotundum (C) bilaterally.
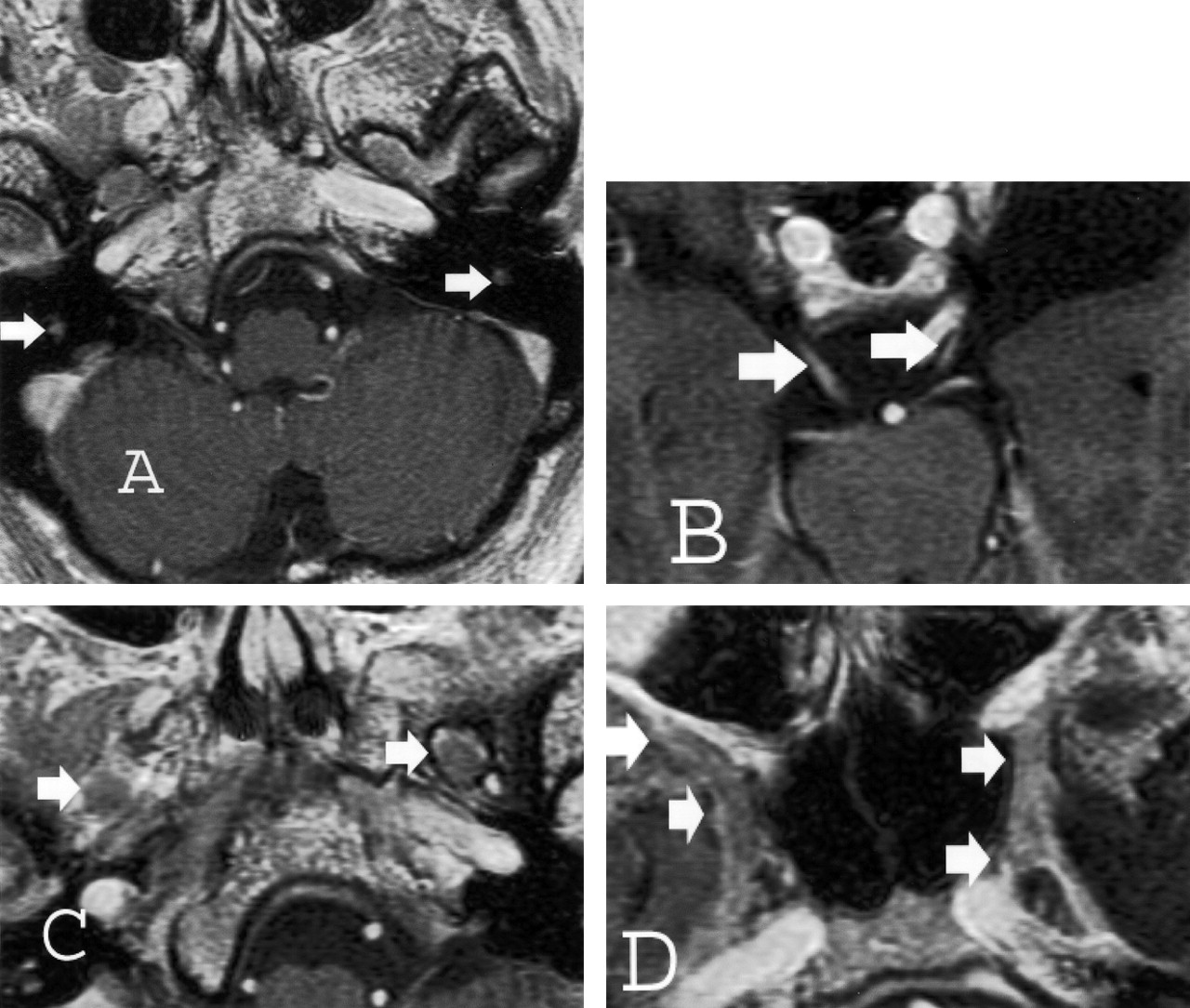
Thin-section (2.5-mm) axial T1-weighted spoiled gradient-echo gadolinium-enhanced images of the brain obtained at 1.5 T for Gamma knife localization (trigeminal neuralgia). Cranial nerve enlargement (arrows) is most pronounced involving the mastoid or descending segments of cranial nerve VII (A), cisternal segments of cranial nerve III (B), V3 (C), and V2 (D) segments. Faint enhancement is identified.
Discussion
The HMSNs represent a group of disorders of various clinical presentations, genetic inheritance patterns, and pathologic and electrophysiologic abnormalities. Dyck et al (7) classified hereditary motor and sensory neuropathies into two main groups based on the presence or absence of decreased nerve conduction velocities and neuronal hypertrophy. HMSN types I, III, and IV, the most common of these disorders, demonstrate both of these abnormalities, but differ in their underlying genetic defects, modes of inheritance, severity of symptoms, and age of onset.
CMT disease represents type I HMSN, a heterogeneous group of disorders of autosomal dominant inheritance characterized by distal muscle weakness, hypoactive or absent tendon reflexes, significantly decreased motor nerve conduction velocities, and hypertrophic onion-bulb changes on nerve biopsy. Symptoms usually begin in the second decade. DNA linkage analysis has demonstrated associations with the Duffy blood antigens on chromosome 1, as well as abnormal loci on chromosome 17 and the X chromosome (forms with linkage to the Duffy group are termed type Ib, others are more frequent and termed type Ia) (1).
Case reports describing the imaging appearance of nerve hypertrophy in this disorder can be found from the mid-1970s with myelography, in the mid-1980s with CT, and to the current time with MR (2–5). Most of these reports pertain to patients presenting with myelopathy, cauda equina, or conus medullaris syndromes secondary to significant nerve hypertrophy within the confines of the central canal. Many of these reports also describe nerve root enlargement in the cervical and thoracic levels concomitantly. Enhancement of the hypertrophied roots is variably present, with faint, if any, enhancement.
Case reports concerning the involvement of cranial nerves in this disorder are rare, although clinical experience has demonstrated an association between CMT and trigeminal neuralgia of uncertain etiology and most often associated with normal nerve size (8). A case report by Shizuka et al (9) demonstrated thickening of the cisternal trigeminal nerves bilaterally on MR images in a patient with CMT. Interestingly, this patient had no clinical symptoms referable to these nerves. We found no other reports concerning the imaging of hypertrophied cranial nerves in this disorder in the literature.
The differential diagnosis of nerve hypertrophy, with or without enhancement, includes such disorders as acute and chronic inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome and CIDP, respectively), other forms of HMSN, neurofibromatosis type 1 (NF-1), neurosarcoidosis, neoplastic disease (metastatic disease and lymphoma), amyloid neuropathy, and neurosyphilis. The degree of cranial nerve enhancement, smooth versus irregular nerve enlargement, and the presence or absence of associated leptomeningeal disease may help in narrowing the differential.
Clinical cranial nerve involvement in the inflammatory demyelinating polyneuropathies is commonly seen; some estimates of cranial nerve symptomatology in CIDP are as high as 50%. Intracranial imaging in patients with Guillain-Barré syndrome is most often normal. A higher proportion of patients with CIDP show intracranial abnormalities—not within the nerves themselves, but within the cerebral white matter analogous to lesions of multiple sclerosis (10).
The frequency of cranial nerve hypertrophy on images in these disorders is substantially lower than that of intracerebral abnormalities. A case report by Duarte et al (11) in 1999 demonstrated significant enlargement of the orbital segments of the oculomotor nerves, as well as enlargement of all three divisions of the trigeminal nerves bilaterally on MR images in a patient with CIDP. Enhancement was identified in all hypertrophied segments, and the trigeminal foramina at the skull base were enlarged. An additional case report by Schady et al (12) in 1996 describes a patient with CIDP who had unilateral deafness and was undergoing surgical biopsy of an inner ear mass, which was subsequently found to represent an enlarged facial nerve. No imaging findings were provided.
Differentiating one type of HMSN from another can be performed by obtaining the history (age at presentation, severity of symptoms, affected relatives, etc), by analysis of nerve conduction velocities, and more recently with genetic analysis. As with CMT, cranial nerve involvement in the other types of HMSN (such as Déjérine-Sottas disease [type III] or Refsum disease [type IV]) is not well described clinically or with imaging.
Additional possibilities in the differential diagnosis are nearly always associated with intense contrast enhancement and/or nodular nerve hypertrophy, decreasing their likelihood. Neurofibromatosis is associated with nerve enlargement secondary to the presence of isolated or plexiform neurofibromas, usually giving a more nodular appearance. Contrast enhancement, especially in plexiform neurofibromas, is variable. A case report by Roos et al (13) describes a patient with a clinical history and imaging features of NF-1 (in the spine), as well as with decreased nerve conduction velocities in the lower extremites. Secondary to this, they questioned concomitant NF-1 and CMT, although pathologic findings of the latter were not present. The genetic diagnosis of CMT was not made, which was likely not an option at the time. The possibility of this seems low, given the lack of other confirmatory evidence. The finding of genetic loci for CMT on chromosome 17, the chromosome of involvement in NF-1, makes this an interesting thought.
Neurosarcoidosis can produce nerve enlargement, which is also typically nodular in appearance with significant enhancement. Leptomeningeal carcinomatosis, from peripheral carcinoma or lymphoma, can produce significant cranial nerve enhancement typically without significant nerve hypertrophy. Neurosyphilis can manifest with cranial nerve enhancement; however, nerve enlargement is not common. Although these disorders can cause foraminal enlargement, this is a rarity. In addition, all three of these disorders are also commonly associated with significant thickening and/or enhancement of the leptomeninges.
Amyloid polyneuropathy, particularly in the lumbar spine, can produce significant nerve root enlargement (14). Intracranially, however, amyloid most commonly manifests as a focal amyloidoma—typically in the region of the gasserian ganglion (15). Cranial nerve enlargement has not been described in this disorder.
Conclusion
The findings in this case, although not specific for the diagnosis of CMT, are consistent with imaging findings of this disorder well described in the lumbar spine. The presence of smooth cranial nerve hypertrophy, absent or faint enhancement, and the absence of associated leptomenigeal disease are important observations that may help exclude other processes in the differential. Although these findings can also be seen in rare cases of CIDP, clinical history, electromyographic findings, and family history are often sufficient to make the distinction between these two disorders.
Acknowledgments
The authors would like to thank the members of the Barrow Neurological Institute Neuromuscular Disease Center for performing the genetic analysis.
References
- Received April 2, 2003.
- Accepted after revision May 25, 2003.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}