
Abstract
SUMMARY: Extraosseous chondroblastoma had been reported in different parts of the body but not intracranially. We report a case of a pathologically proven intracranial extraosseous chondroblastoma of the right cavernous sinus in an 18-year-old woman with CT, MR, and conventional angiographic features simulating meningioma.
Chondroblastoma is an uncommon benign cartilaginous bone tumor that typically occurs at the epiphysis of the long bones in the first 2 decades of life. When it occurs in older patients, chondroblastoma tends to involve unusual sites, such as the skull and pelvis.1,2 Although most chondroblastoma remains nonaggressive and has a benign course, local recurrence, malignant transformation, and distant metastasis have all been documented.3–6 Few cases of osseous intracranial chondroblastoma involving the cranial bones have been reported.1,2,7–10 We report the first case of extraosseous chondroblastoma occurring intracranially. The imaging features on CT, MR, and conventional cerebral angiography were similar to meningioma.
Case Report
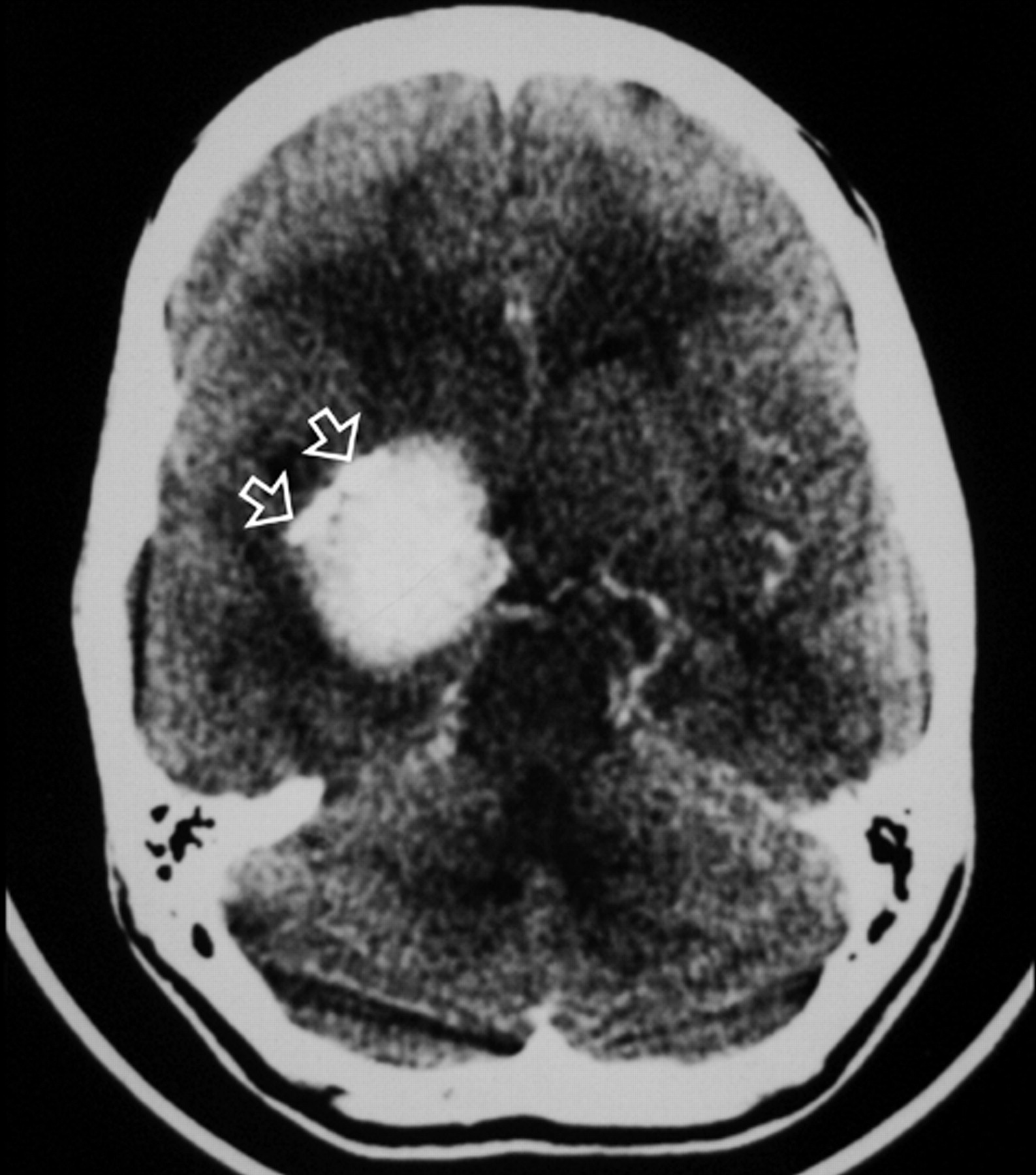
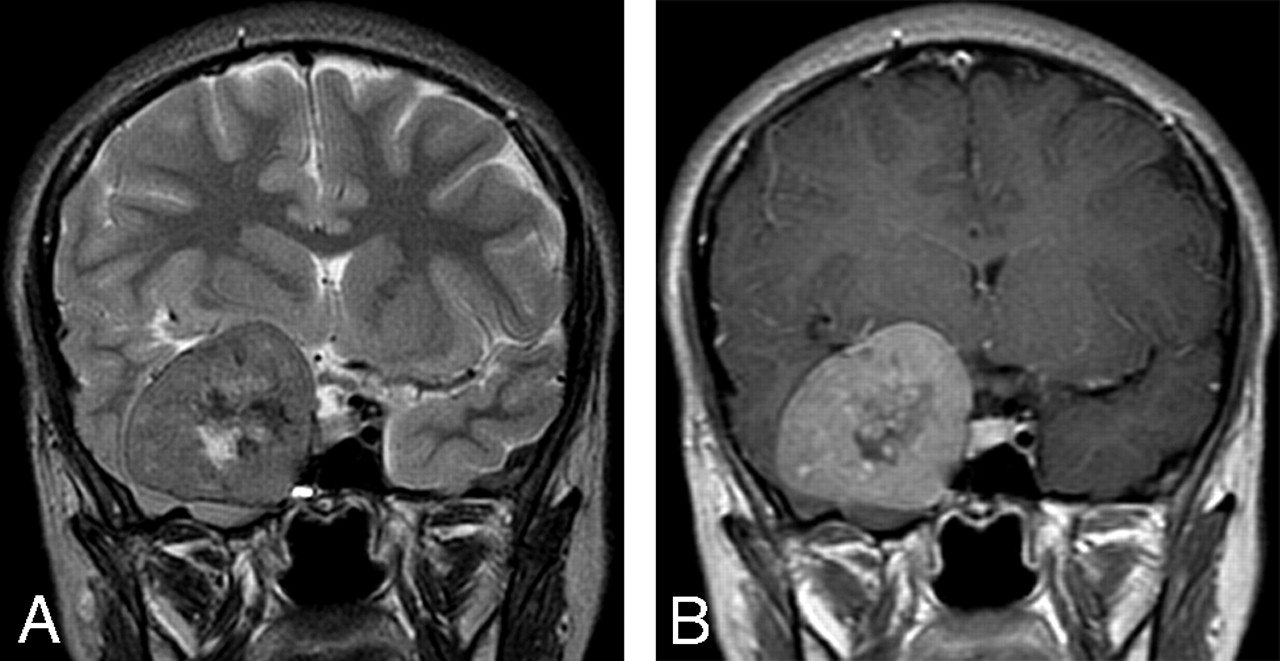
An 18-year-old woman was referred to our institution with a 4-year history of generalized headache and 6-month history of pain, blurring of vision, diplopia, and gradual proptosis of the right eye. Past medical history was unremarkable. Neurologic examination revealed complete right third and fourth cranial nerve palsy with partial sixth-nerve palsy. The right pupil was dilated, fixed, and not reacting to light or accommodation directly or consensually. Funduscopic examination revealed blurring of the medial aspect of the right optic disc and normal left optic disc. Contrast-enhanced brain CT examination showed a large enhancing right parasellar extra-axial mass with peripheral calcification but no associated bone involvement (Fig 1). Enhanced brain MR examination revealed a mass (4 × 5 × 4 cm) arising from the lateral wall of the right cavernous sinus encasing and displacing the right internal carotid artery (ICA) medially and anteriorly. The mass had extended into the right optic canal and compressed the right optic nerve. On T2-weighted images, the mass demonstrated predominantly intermediate signal intensity with the center showing mixed low and high signal intensity (Fig 2A). On T1-weighted images, the mass demonstrated intermediate signal intensity with strong and homogenous enhancement, except for the center, which failed to enhance (Fig 2B). Small enhancing dural tail was seen at the attachment with the dura. Signal intensity void areas at the periphery of the mass were present corresponding with the calcifications seen on the CT. A conventional cerebral angiogram showed tumor blush in the right parasellar region in the early arterial phase with appearance similar to sunburst and late washout (Fig 3). The tumor blood supply was derived mainly from the cavernous segment of the right ICA. The mass had compressed and narrowed the cavernous and suprasellar segments of the right ICA. The supraclinoid segments of the right ICA and the right middle cerebral artery were displaced posteriorly. A small nipple-shaped, wide neck aneurysm arising from the anterior aspect of the supraclinoid segment of the right ICA was present. Branches of the right external carotid artery, mainly the middle meningeal artery, also contributed to the blood supply of the tumor.

Axial contrast-enhanced CT image of the brain just above the level of the suprasellar cistern showing intensely enhancing right-sided mass with peripheral calcifications (arrow).

Coronal T2-weighted (A) and contrast-enhanced T1-weighted (B) MR images at the level of sella turcica demonstrating the relation of the lesion to the right cavernous sinus. The intermediate signal intensity of the lesion on T2-weighted images and the pattern of enhancement are very suggestive of meningioma.

Anteroposterior projection of right ICA arteriogram. A, Strong tumor blush is seen in the right parasellar region during the early arterial phase with presence of feeding vascular pedicle arising from the cavernous segment of ICA and sun ray pattern of tumor blush. The tumor is compressing and displacing the right ICA and middle cerebral artery. B, Persistent tumor blush in the late venous phase.
A preoperative diagnosis of meningioma was made, and right pterional craniotomy was performed. At surgery, a heavily calcified hypervascular mass was found in the right middle cranial fossa adjacent to the greater wing of the sphenoid filling the right cavernous sinus. Almost total macroscopic excision was achieved, and the aneurysm was wrapped with muscle and tissue. The patient had slow but steady uneventful postoperative recovery. There was no new focal neurologic deficit apart from her known preoperative third, fourth, and sixth cranial nerves palsies. The histopathologic examination of the tumor specimen revealed numerous multinucleated giant cells in a background of early calcifying chondroblasts; some of them showing grooved nuclei (Fig. 4). The findings were consistent with chondroblastoma. On follow-up 3 months after surgery, the patient's right orbital pain and diplopia had improved, and the clinical examination showed complete recovery of the fourth and sixth cranial nerve palsies with persistent third cranial nerve palsy.

Photomicrograph shows highly cellular chondroid matrix with numerous chondroblast cells, giant cells, and mononuclear cells (hematoxylin-eosin, ×200).
Discussion
In 1928, Ewing first described the calcifying giant cell tumor, subsequently designated “epiphyseal chondromatous giant cell tumor” by Codman in 1931.11 The term chondroblastoma was introduced by Jaffe and Lichtenstein in 1942.12 Although well recognized, chondroblastoma is still a rare tumor accounting for less than 1% of all bone tumors.13 It involves the epiphysis of long bones in 97% of cases,2 predominantly occurs in the first 2 decades of life, and boys are affected twice as often as girls.5,7,13 Approximately 83% of chondroblastomas of the skull and face occur in patients older than 30 years, whereas 92% of chondroblastomas in long bones occur in patients younger than 30 years of age.14,15 When chondroblastoma occurs in older patients, it tends to involve unusual sites like the skull and facial bones.14 The most common location of chondroblastoma involving cranial bones is the squamous portion of the temporal bone accounting for 70% of the cases.7,8 Extraosseous chondroblastomas had been reported in the soft tissue of different parts of the body, but no previous reports had documented intracranial extraosseous involvement. Most chondroblastomas remain nonaggressive and have a benign course. However, local recurrence, malignant transformation to sarcoma (often after radiation therapy), and distant metastasis have all been reported.3–6
Radiographically, chondroblastoma in the long bones is characterized by a well-defined osteolytic lesion that involves an epiphysis or secondary ossification center, often with foci of mottled calcification.7,16 Intracranial chondroblastoma involving the skull bones has been reported to have a fuzzily rarefied appearance and sharp demarcation from the adjacent bone by a thin margin of increased attenuation.9,10,17 CT scan confirms the lytic nature of the lesion and shows areas of calcifications in the center and the periphery of the tumor. MR imaging demonstrates variable patterns. The tumor can show hypointensity on T1-weighted images, hyperintensity on T2-weighted images, and marked contrast enhancement1 or it can show heterogenous solid and multilobulated cystic components with low signal intensity of the solid component on both T1- and T2-weighted images and fluid-fluid levels of the cystic component with marked enhancement in the solid component and the septa of the cystic part.7
Our report is the first report of an intracranial extraosseous chondroblastoma. The imaging findings on CT, MR imaging, and conventional angiography were very similar to meningioma. The preoperative diagnosis of meningioma in our patient was based on the extra-axial and extraosseous location of the tumor and its relation to the dura, as well as the signal intensity and pattern of enhancement on MR. The angiogram findings of strong tumor blush with vascular pedicle supplying the tumor in a radial pattern and delayed washout further supported the diagnosis of meningioma; however, the histopathologic examination eventually confirmed the diagnosis of chondroblastoma.
References
- Received May 9, 2007.
- Accepted after revision May 24, 2007.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.