
Abstract
BACKGROUND AND PURPOSE: Sturge-Weber syndrome (SWS) is frequently associated with neurologic complications such as seizures, so diagnosing this condition has important implications for patient management. The purpose of this study was to report unusual neuroimaging findings in patients with facial port-wine stain (PWS) and clinically suspected SWS.
MATERIALS AND METHODS: Cranial MR imaging was reviewed for all children with facial port-wine stain (PWS) involving the upper face and eyelids who were referred to Great Ormond Street Hospital between 2003 and 2007 for investigation of suspected SWS. Patients were excluded from further analysis if the imaging findings were normal on initial and subsequent scans and the subject remained free of neurologic disease, or if the imaging showed the well-recognized pattern of exclusively supratentorial pial enhancement representing the pial angioma of SWS. For the remaining patients, the neurologic, dermatologic, and ophthalmologic records were examined and all available imaging was reviewed by a neuroradiologist. We documented the presence and distribution of pial enhancement; corroborative features of SWS, such as atrophy, calcification, choroid plexus changes, and ocular abnormalities; and all other intracranial abnormalities.
RESULTS: Of the 62 patients referred for assessment, imaging findings were considered typical of SWS in 32 (52%) and were normal or showed abnormalities attributable to an unrelated pathology in 20 (32%). Of the remaining 10 patients, in 7 (11%), there was evidence of a pial angioma in an unusual distribution involving infratentorial structures, with the angioma in 1 patient being diagnosed at postmortem only; in 2 (3%), there were imaging abnormalities with some features in common with typical SWS, such as subcortical calcification, but with no evidence of pial enhancement; in 1 (1.6%), the initial MR imaging finding was normal, but repeat imaging subsequently revealed pial enhancement.
CONCLUSIONS: Involvement of infratentorial structures is common but may be relatively subtle and should be actively sought. Cases in which there are certain patterns of imaging abnormalities but an apparent absence of supratentorial pial enhancement on MR imaging may represent formes frustes of SWS; visualization of pial angiomatosis may also be delayed until later in childhood than expected.
Sturge-Weber syndrome (SWS) is a neurocutaneous disorder characterized by angiomatosis of the skin, eye, and meninges. The expression of the syndrome is variable1 but typically manifests as facial port-wine stain (PWS), ocular choroidal hemangiomas, and cerebral pial angiomatosis. Glaucoma and neurologic complications, including seizures and developmental delay, are common,1,2 so diagnosing SWS has important implications for patient management. The original description by Sturge in 18793 was of the association of a facial PWS, glaucoma, and focal seizures. Subsequently Weber 4 described the radiologic correlates of pial angiomatosis, demonstrating involvement of the brain. The current working definition of SWS has not been formally defined but usually depends on the presence of a pial angioma involving the cerebral cortex, with or without neurologic manifestations.
Although a facial PWS is almost always present in cases of SWS, it is not synonymous with the diagnosis, and in most instances, facial PWS is an isolated uncomplicated finding. Only those patients with PWS affecting the upper face and eyelids, approximating the sensory distribution of the ophthalmic division of the trigeminal nerve (V1), are considered at risk of the neuro-ophthalmologic complications characteristic of SWS,5,6 and recognition of a PWS in this distribution is the usual trigger for further investigation. Even within this selected group, only 26% go on to develop such complications,6 and the diagnostic challenge is in recognizing these patients as early as possible, preferably before the onset of neuro-ophthalmologic sequelae. The typical neuroimaging findings of SWS are well described,7,8 and cross-sectional imaging, usually with MR imaging, plays a pivotal role in establishing the diagnosis.
The purpose of this study was to report unusual neuroimaging findings in patients with facial PWS and clinically suspected SWS.
Materials and Methods
This work was approved by the Great Ormond Street Hospital research ethics committee.
The cohort for the study comprised all patients referred to Great Ormond Street Hospital Department of Neuroradiology between 2003 and 2007 for imaging assessment of suspected SWS. The criterion for referral was the presence of a facial PWS involving, but not necessarily restricted to, the distribution of V1. All patients had 1 or more MR imaging brain examinations, which included gadolinium-enhanced T1-weighted (T1WI) sequences. For MR imaging examinations performed within our institution, a standardized brain imaging protocol was applied by using a 1.5T scanner (Symphony or Avanto; Siemens, Erlangen, Germany). Studies performed before 24 months of age comprised axial dual-echo short tau-inversion-time (STIR) inversion recovery with T1WI axial, coronal, and sagittal sequences post-gadopentate dimeglumine. For studies performed after 24 months of age, the dual-echo was replaced by an axial T2-weighted (T2WI) sequence. Patients were excluded from further consideration if initial and subsequent imaging findings were normal and the subject remained free of neurologic disease, if imaging demonstrated an abnormality readily attributable to a pathology other than SWS, or if the first MR imaging demonstrated findings typical of SWS. For the purposes of this report, the essential criterion for the diagnosis of typical SWS was considered to be the presence of pial enhancement isolated to the supratentorial compartment, representing the presence of a pial angioma.
For the remaining patients, the MR imaging was reviewed again, along with any supportive imaging such as CT scans, by a neuroradiologist (M.E.A.). In particular, the presence and distribution of pial enhancement; corroborative features of SWS such as atrophy, calcification, choroid plexus changes, and ocular and venous abnormalities; and all other intracranial abnormalities were documented. The clinical notes were reviewed to obtain the details of neurologic or ophthalmologic abnormalities, and where possible, clinical photography was reviewed to determine the distribution of the facial PWS. Due to easy reproducibility and widespread use within the literature, the topography of the PWS was classified according to the sensory dermatomal territories of the trigeminal nerve (ophthalmic division [V1], maxillary division [V2], and mandibular division [V3]).
Results
During the period 2003–2007, sixty-two cases of clinically suspected SWS were referred for imaging assessment. The median age at the time of the first MR imaging was 11.5 months (range, 1–168 months). Findings were considered typical of SWS on imaging in 32 (52%) subjects, and they were normal on imaging and clinical examination or showed abnormalities attributable to an unrelated pathology in 20 (32%) subjects. The remaining 10 patients (16%) were included for further analysis. Nine of the 10 patients had clinical photographs of the facial PWS available for review. Patient demographics, clinical features, and imaging findings are summarized in the on-line Table. The following subheadings summarize these findings.
Infratentorial Pial Angioma
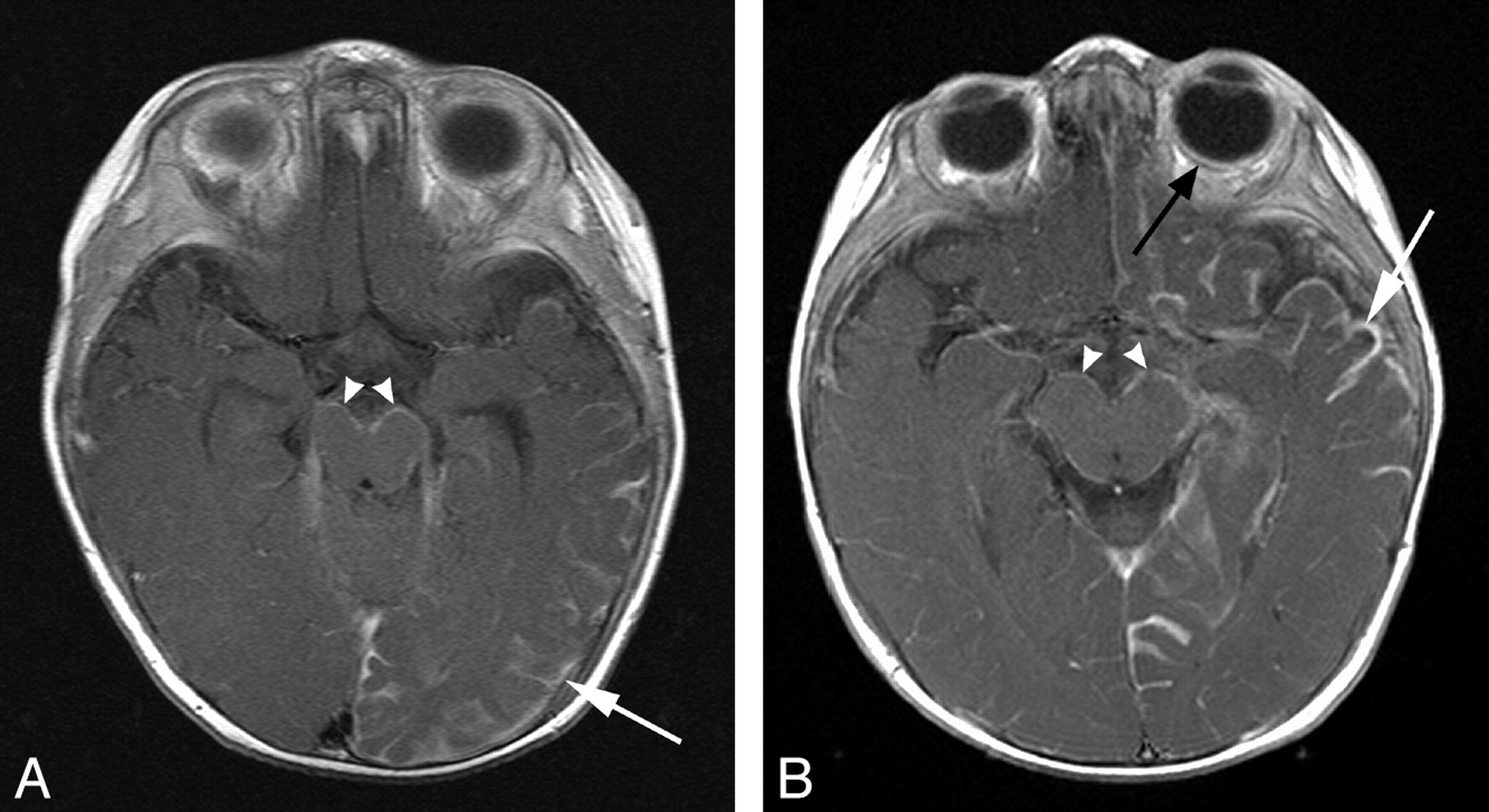
Of these 10 patients, in 7 (11%), there was evidence of a pial angioma involving infratentorial structures. Three (43%) had involvement of both supra- and infratentorial structures, and 4 (57%) had an angioma isolated to the brain stem and cerebellum. Within the subgroup with both supra- and infratentorial involvement, there were 2 patients in whom MR imaging demonstrated cerebral hemispheric involvement with bilateral midbrain and pontine pial enhancement (Fig 1). The third patient in this subgroup had pial enhancement and progressive atrophy of the right cerebral hemisphere and contralateral left cerebellar hemisphere (Fig 2). Of the 4 patients with isolated infratentorial pial angioma (Fig 3), 1 case was not recognized on serial MR imaging or CT but was diagnosed at postmortem examination (Fig 4). This subject had continued to remain under surveillance because of clinical concerns, despite the lack of supportive imaging.
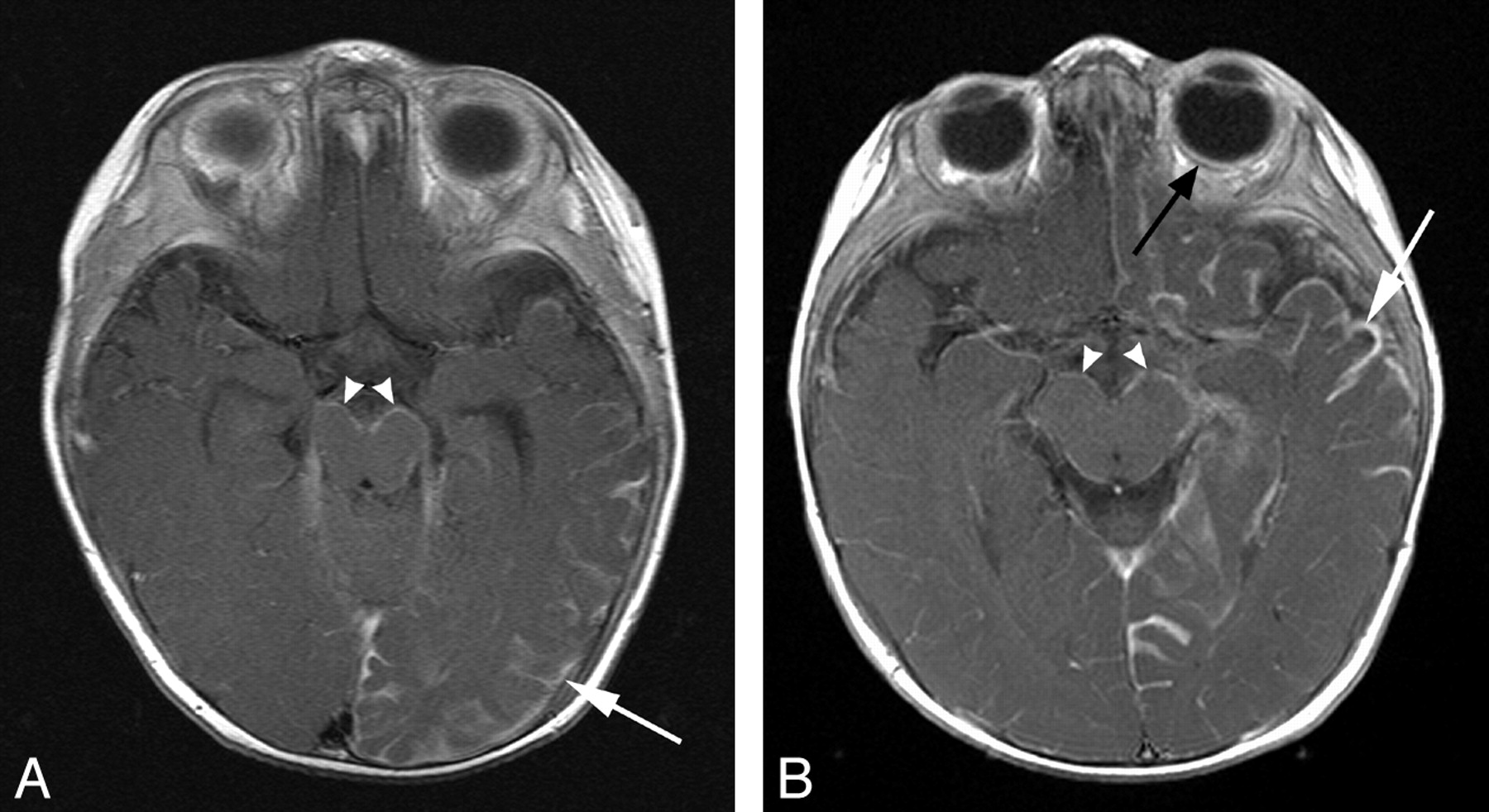
Pial angioma involving both supra- and infratentorial compartments. A and B, T1WI postgadolinium MR images of different patients (8 and 5 months of age, respectively) demonstrate pial enhancement of the left cerebral hemisphere (white arrows) and midbrain bilaterally (arrowheads). Choroidal angiomas (black arrow) and choroid plexus hypertrophy were also evident localized to the side of supratentorial involvement.

Coronal MR images of an 8-month-old boy. A, T2WI image demonstrates atrophy of the right cerebral hemisphere and left cerebellar hemisphere. B, T1WI postgadolinium image demonstrates pial enhancement of the right cerebral hemisphere (white arrows) and left cerebellar hemisphere. C, Magnified T1WI postgadolinium image of the left cerebellar hemisphere demonstrates enhancement of the pial surface of the folia (white arrows). Bilateral midbrain pial enhancement and choroid plexus hypertrophy were also evident in this case.

Pial angioma involving the infratentorial compartment only. A, Axial T1WI postgadolinium image of a 12-month-old boy demonstrates pial enhancement of the left cerebellar hemisphere (arrows) and medulla bilaterally (arrowheads). B, Axial T1WI postgadolinium image demonstrates pial enhancement of the ventral pons (arrowhead) and interpeduncular region of the midbrain (inset, arrowhead).
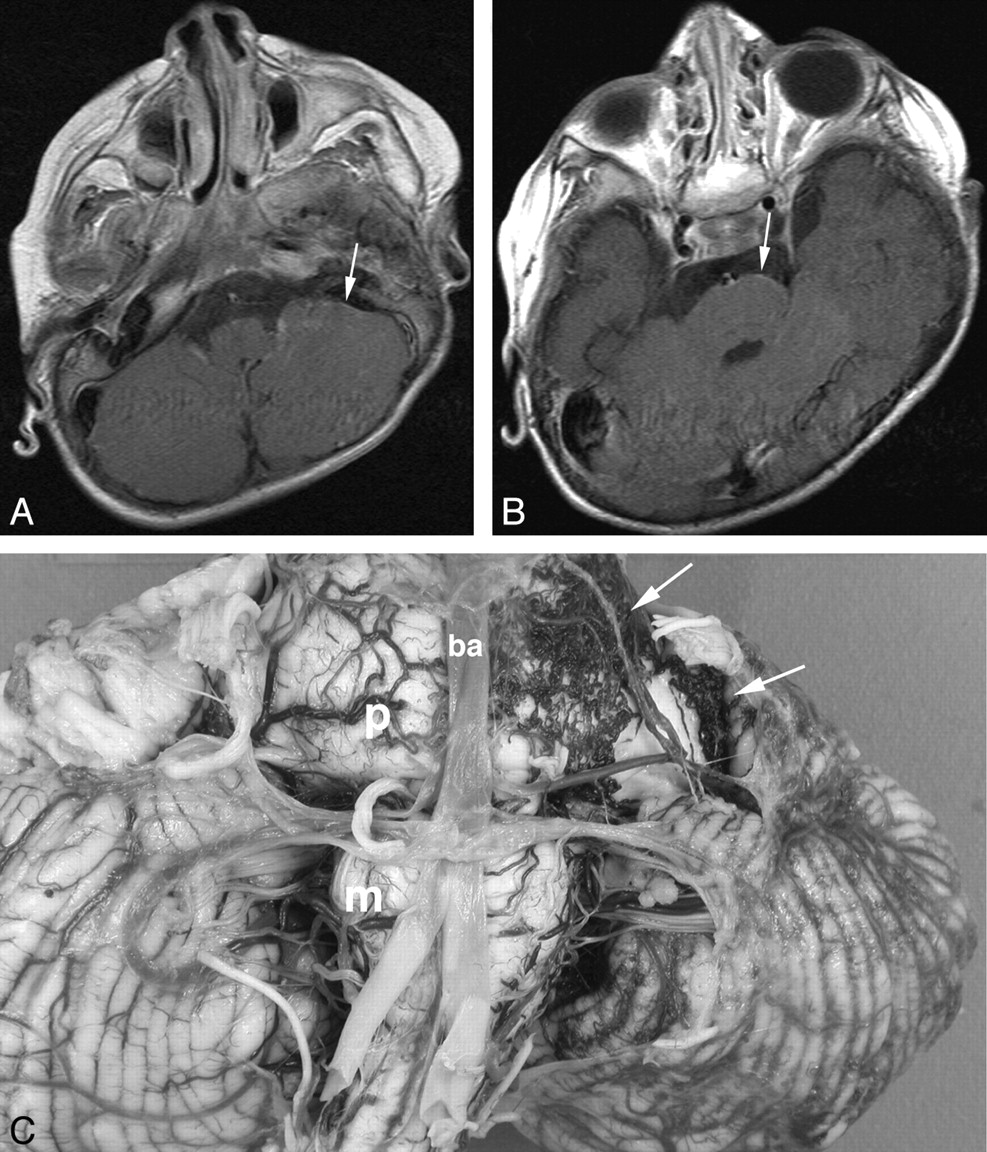
Female patient with left-sided facial PWS isolated to V1 and V2, who developed early-onset seizures and developmental delay and died at 6 years of age secondary to pneumonia. Initial MR imaging at 6 months of age (not shown) demonstrated postural plagiocephaly and mild global delay in myelination, but no features of SWS. A and B, Axial T1WI postgadolinium MR images at 32 months of age. At the time of the examination, the images were interpreted as showing no features of SWS. Retrospectively, possible subtle pial enhancement is seen overlying the left pons and cerebellar hemisphere (arrows). C, Postmortem gross pathologic specimen of the cerebellum and brain stem viewed from the ventral surface. A fine plexus of abnormal vessels is seen overlying the left side of the pons, medulla, and left cerebellar hemisphere (arrows). P indicates pons; m, medulla; ba, basilar artery.
Of these 7 patients with infratentorial pial angioma, 5 (71%) had a PWS involving all 3 territories of the trigeminal nerve and 2 had PWS involving V1 and V2 but sparing V3. Two of the 7 patients (29%) developed seizures, 2 (29%) had generalized developmental delay, and 1 (14%) exhibited slightly delayed gross motor development.
SWS without Pial Enhancement
There were 2 cases (3% overall) with no pial enhancement but with additional MR imaging abnormalities and clinical findings providing compelling evidence for a diagnosis of SWS, without histologic or postmortem concordance. One patient had imaging findings of progressive macrocrania. Clinically, the patient had increasing head circumference from infancy, plateauing at 4 years of age, and right glaucoma. MR images obtained at 6, 16, and 45 months demonstrated fine vessels within the subarachnoid spaces bilaterally, progressive macrocrania, and enlargement of the right globe. MR venography used to supplement the final study confirmed patency of the dural venous sinuses and deep venous system with no clear abnormality of the superficial cortical venous system. The second patient had a clinical complex of early-onset severe seizures and fatally progressive encephalopathy. Serial CT and MR imaging demonstrated progressive atrophy and subcortical calcification over 15 months (Fig 5).

Serial imaging of a child with a clinical complex of bilateral facial PWS, early-onset severe seizures, and fatally progressive encephalopathy. Imaging demonstrates progressive cerebral atrophy and calcification without evidence of pial enhancement. A, Contrast-enhanced CT scan at 1 month of age demonstrates mild cerebral atrophy and bilateral choroid plexus hypertrophy (black arrow). B, Axial T1WI postgadolinium image at 12 months of age shows further cerebral volume loss but no evidence of abnormal pial enhancement. C, CT scan at 15 months of age demonstrates progressive symmetric cerebral volume loss with calcification of the cortex and subcortical white matter, most marked within the frontal lobes bilaterally (white arrows).
Delayed Visualization of Pial Enhancement
In 1 patient (1.6% overall), pial enhancement was not present on the initial scan but was confirmed only on subsequent follow-up imaging. At first presentation, the patient had no neurologic problems. The initial MR imaging at 24 months of age demonstrated plethoric deep medullary and subependymal veins bilaterally, with multiple developmental venous anomalies (DVAs), an enlarged left globe, but no evidence of pial angiomatosis. At 56 months of age, she developed a gradual onset of aphasia and right-sided hemiplegia, which proceeded to complete recovery within 3 days. There was no clinical evidence of seizure activity during this period. Further MR imaging performed 1 week after resolution of symptoms demonstrated new abnormal leptomeningeal enhancement overlying the parieto-occipital regions bilaterally. In the left parieto-occipital region, where the abnormal enhancement was most conspicuous, there was also subtle loss of parenchymal volume and hypointensity within the subcortical white matter on T2WI sequences.
Discussion
Although the exact etiology of SWS remains unclear, the fundamental pathophysiologic abnormality in areas of brain affected by pial angiomatosis is the absence of an adequately functioning superficial cortical venous system.7 Consequently, blood is rerouted centrally via the medullary veins resulting in venous hypertension and stagnation.9 The typical imaging features of SWS reflect the presence of angiomatosis and the sequelae of the related hemodynamic disturbances. MR imaging is currently the imaging investigation of choice, with pial enhancement on postgadolinium sequences considered the gold standard in assessing the extent of disease7 and regarded by some as being the most important criterion in the radiologic diagnosis.10 Pial enhancement characteristically involves the occipital and posterior parietal cortex ipsilateral to the PWS,7,11 though involvement of the frontal and temporal lobes is also well recognized.7,10
Making the diagnosis of SWS, preferably before the onset of neurologic sequelae, may have an important impact on patient management. Aggressive treatment of epilepsy is advocated,12 with some studies even suggesting neurodevelopmental benefits with prophylactic anticonvulsants before the onset of seizures.13 Prophylactic daily aspirin14 and early surgery when seizures are not controlled by medical treatment may also be of benefit.12 SWS has a spectrum of clinical and imaging findings, and it is important to recognize those patients at the fringes of the spectrum who may benefit from therapies and monitoring applied in more conventional cases. In this study, we identified patients constituting 16% of all those referred with suspected SWS in whom imaging findings were not typical but who might represent such an extended spectrum of SWS.
Posterior fossa involvement is a rare finding in SWS, and reported cases are exceedingly scarce.15 In this series, we presented a total of 7 cases, 4 of which had pial angiomatosis isolated to the infratentorial compartment. In 1 case, the pial angioma was not recognized on contrast-enhanced MR imaging. In 2 others, the infratentorial imaging findings were of pial enhancement only. The relative subtlety of the imaging appearances means that involvement of infratentorial structures may be easily overlooked if not actively sought, and we suggest that radiologists should regard this as an important review area when reporting the findings of scans of patients with suspected SWS.
Of particular interest in the group of patients with involvement of the infratentorial compartment was the case of pial enhancement and progressive atrophy of the right cerebral hemisphere and contralateral cerebellar hemisphere. The volume loss of the opposite cerebellar hemisphere could be at least in part explained by the phenomenon of crossed cerebellar diaschisis, in which functional disconnection of the cerebral cortex results in hypometabolism of the contralateral cerebellar hemisphere. Cerebellar volume loss is described in crossed cerebellar diaschisis16; and Yoshikawa et al17 reported a child with SWS in whom there was a reduction in the cerebral blood flow and metabolism in the cerebral cortex underlying the angioma and in the contralateral cerebellum. However, to our knowledge, folial pial enhancement has not been described. In the case reported here, the presence of leptomeningeal angiomatosis with parenchymal atrophy secondary to the same venous hypertension proposed for supratentorial changes offers the most likely explanation for the imaging appearances. The distribution of bilateral PWS in this case involved all 3 dermatomes of the trigeminal nerve, raising questions regarding the mechanisms by which the phenotype of SWS is expressed, particularly in the context of those theories proposing an insult to groups of cells in close proximity to one another in the developing embryo.18
Recently, it has been proposed that the proximity of pluripotent neural crest cells within primitive embryologic segments known as metameres accounts for the distribution of cutaneous and neurovascular manifestations of several syndromes, including SWS.18 In this model, nevi of the mandible are associated with involvement of rhombencephalic structures. In our group of patients with infratentorial pial angiomatosis, 2 of 7 (29%) did not have PWS affecting V3, and in a further patient, V3 was involved only by virtue of PWS affecting the pinna, with no nevus evident overlying the mandible itself. Such observations would not seem to support this metameric theory, though our overall cohort of patients was biased in this respect because it did not examine those in whom the PWS was isolated to V3.
Of the patients with pial angioma isolated to the infratentorial compartment, there was 1 patient with mild motor delay and 1 patient with seizures and developmental delay. The latter patient also had generalized white matter volume loss and signal-intensity abnormality, speculatively due to an as-yet-undiagnosed second pathology. The clinical importance of recognizing pial angiomatosis isolated to the infratentorial compartment will be the subject of long-term follow-up.
With regard to the case of progressive macrocephaly without radiologic evidence of pial angioma, 5 cases with similar imaging findings have been described in the literature.19–22 In all of these cases, there were concomitant abnormalities of either the deep venous system or jugular outflow. As a result, venous hypertension was universally the proposed mechanism for the abnormal enlargement of the brain parenchyma and/or ventricular system. In the case reported here, MR venography demonstrated normal major deep veins and dural sinuses. Macrocrania in this instance might reflect a disturbance of venous hemodynamics at a point other than the major deep draining veins and dural sinuses. If this were the case, the venous abnormalities reported in other patients, such as occlusion of the deep venous system, might represent a sequel of chronic venous hypertension rather than a causative agent. Although it is possible that our patient simply had a PWS and a separate coincidental pathology such as benign macrocrania of infancy, the presence of supportive features of SWS such as glaucoma mitigates against this.
A case with imaging findings of progressive atrophy and calcification but with no radiologic evidence of pial angiomatosis has previously been described,23 with the findings being attributed to regression of the angioma secondary to thrombosis. A separate report described reduction of leptomeningeal enhancement with time with a concomitant increase in the prominence of the deep venous system.10 It is also recognized that in cases of burnt-out SWS, once the cortex has been reduced to a nonfunctioning poorly perfused calcified mantle, the associated abnormal leptomeningeal vessels regress due a lack of capillary runoff.24
Our 2 cases in which pial angioma was recognized only on delayed imaging or at postmortem serve to illustrate that initial imaging may be insensitive to this reputedly cardinal feature of SWS. A study examining the utility of blood oxygen level−dependent (BOLD) imaging in SWS has documented a case in which leptomeningeal enhancement was absent on initial MR imaging at 4 months of age but became evident on repeat imaging 8 months later.25 Balselga 1 recognized that pial angiomatosis may be difficult to identify in the neonatal period and advocated repeat imaging at 12 months if initial studies are nondiagnostic, postulating that increased dilation of meningeal vessels with time leads to improved recognition. However, in our cases, MR imaging performed at 33 months and 25 months of age, respectively, did not demonstrate a pial angioma, raising questions as to the optimum time for imaging and the need for serial scanning in high-risk individuals.
An alternative explanation for the apparent absence of a pial angioma on imaging in 3 cases and delayed visualization in 1 case is that the imaging techniques used were not sensitive enough. The leptomeningeal enhancement seen in SWS is thought to reflect either slow turbulent flow through the tortuous veins of the pial angioma or leakage of contrast due to a disturbance of the blood-brain barrier.24 It is proposed that repetitive or prolonged seizure activity produces permeability changes in the blood-brain barrier.26 Indeed, several studies describe cases of SWS in which cerebral leptomeningeal enhancement transiently increased immediately following seizure activity,27–29 sometimes with complete resolution on subsequent imaging.29 It is possible that subtle pial angiomas only become evident on conventional contrast-enhanced imaging once the seizure activity or venous ischemia within the underlying cortex results in sufficient disturbance of the blood-brain barrier or venous stasis within the angioma to unmask its presence. This might explain the findings in the case in which demonstration of the supratentorial angioma was delayed, though there was no clinical evidence of seizures and the patient had made a complete recovery at the time of imaging.
Although gadolinium-enhanced T1WI imaging is currently the MR imaging of choice for the detection of pial angiomatosis, recent studies have demonstrated that postcontrast fluid-attenuated inversion recovery sequences30 and BOLD MR venography 25 may provide increased sensitivity. In addition, susceptibility-weighted imaging has been shown to detect abnormalities of the medullary veins and cortical calcification earlier than conventional MR imaging.31 If validated, such imaging techniques may provide useful adjuncts to more conventional MR images in the future.
Conclusions
Prompt diagnosis of SWS is essential so that management targeted at improving long-term neurologic outcomes can be commenced as early as possible. Although the typical imaging findings of SWS are well described, it is clear that a spectrum of appearances exists. Involvement of infratentorial structures is often subtle and should be actively sought. Although demonstration of the pial angioma is often considered crucial for the diagnosis of SWS, visualization on MR imaging may be delayed; the apparent absence of pial enhancement should be interpreted with caution, particularly in the presence of other features of SWS. These observations should be taken into consideration when interpreting imaging and developing strategies for the investigation of suspected SWS.
Footnotes
indicates article with supplemental on-line table.
References
- Received November 11, 2007.
- Accepted after revision September 11, 2008.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}