
Abstract
BACKGROUND AND PURPOSE: Kennedy disease is a rare X-linked neurodegenerative disorder caused by a CAG repeat expansion in the first exon of the androgen-receptor gene. Apart from neurologic signs, this mutation can cause a partial androgen insensitivity syndrome with typical alterations of gonadotropic hormones produced by the pituitary gland. The aim of the present study was therefore to evaluate the impact of Kennedy disease on pituitary gland volume under the hypothesis that endocrinologic changes caused by partial androgen insensitivity may lead to morphologic changes (ie, hypertrophy) of the pituitary gland.
MATERIALS AND METHODS: Pituitary gland volume was measured in sagittal sections of 3D T1-weighted 3T-MR imaging data of 8 patients with genetically proven Kennedy disease and compared with 16 healthy age-matched control subjects by use of Multitracer by a blinded, experienced radiologist. The results were analyzed by a univariant ANOVA with total brain volume as a covariant. Furthermore, correlation and linear regression analyses were performed for pituitary volume, patient age, disease duration, and CAG repeat expansion length. Intraobserver reliability was evaluated by means of the Pearson correlation coefficient.
RESULTS: Pituitary volume was significantly larger in patients with Kennedy disease (636 [±90] mm3) than in healthy control subjects (534 [±91] mm3) (P = .041). There was no significant difference in total brain volume (P = .379). Control subjects showed a significant decrease in volume with age (r = −0.712, P = .002), whereas there was a trend to increasing gland volume in patients with Kennedy disease (r = 0.443, P = .272). Gland volume correlated with CAG repeat expansion length in patients (r = 0.630, P = .047). The correlation coefficient for intraobserver reliability was 0.94 (P < .001).
CONCLUSIONS: Patients with Kennedy disease showed a significantly higher pituitary volume that correlated with the CAG repeat expansion length. This could reflect hypertrophy as the result of elevated gonadotropic hormone secretion caused by the androgen receptor mutation with partial androgen insensitivity.
ABBREVIATIONS:
- KD
- Kennedy disease
- AR
- androgen receptor
- LH
- lutropin
- FSH
- follicle-stimulating hormone
- SHBG
- sex hormone–binding globulin
Spinobulbar muscular atrophy (Kennedy disease [KD]) is a rare X-linked neurodegenerative disorder of mainly spinal and bulbar motoneurons. It is caused by a CAG repeat expansion in the first exon of the androgen receptor (AR). Apart from typical neurologic signs such as a progressive wasting of proximal muscles and bulbar symptoms, this mutation causes a partial androgen insensitivity syndrome in >80% of patients, with gynecomastia being the most prominent symptom.1⇓–3
Androgen insensitivity syndromes are typically associated with normal or elevated testosterone and estrogen levels, with elevated lutropin (LH) and sex hormone–binging globulin (SHBG) levels. This pattern can also be found in patients with KD.3
Battaglia et al4 described a case of a 59-year-old patient with KD who presented with typical endocrine symptoms, but without any apparent physical signs of neurodegeneration.
The pituitary gland is known to produce hormones such as LH, which control the activity of other endocrine glands.
Because of high spatial and contrast resolution, MR imaging is currently the technique best suited to examine the pituitary gland.5⇓⇓–8 The normal size of the gland in a healthy adult is approximately 5–10 mm in height, 10 mm in length, and 10–15 mm in width.9
Pituitary gland volumetry
It is well established that dysfunctions such as primary hypothyroidism leading to elevated thyroid-stimulating hormone levels are associated with an enlargement of the pituitary gland.10
The aim of the present study was to evaluate the impact of KD on the volume of the pituitary gland as measured by MR imaging under the hypothesis that the described changes in gonadotropic hormone levels in KD can lead to morphologic changes, namely, hypertrophy of the pituitary gland. To the best of our knowledge, this is the first study to examine the volume of the pituitary gland in patients with KD.
Materials and Methods
Patients
The patient group consisted of 8 men (age, 44–73 years; mean, 53.8) with genetically proven KD with no neurologic comorbidity. The control group included 16 age-matched healthy men (42–77 years; mean, 54.4). Informed consent was obtained from each subject. The study was approved by the local ethics committee.
All patients were clinically examined at the Department of Neurology.
All 8 patients showed typical symptoms of KD reflecting neurodegeneration. Disease duration ranged from 2–19 years (mean, 10.5 ± 6.2 years). Muscle weakness was present in 100% to different degrees. Muscular atrophy and fasciculations were most prominent in the tongue in 6 of 8 and 7 of 8 patients, respectively. All patients had dysphagia and 4 of 8 had dysarthria, as signs of bulbar neuron involvement.
Signs of partial androgen insensitivity were present in all patients. Six of 8 presented with severe gynecomastia. One patient had erectile dysfunction, and 4 of 8 fathered children. Hormone levels were determined in 2 patients previous to the first MR imaging examination, which showed elevated testosterone of 12.47 ng/mL (normal values <7.99 ng/mL) and SHBG in one patient and values at the upper limit of the normal range for testosterone and LH in the other.
Data Acquisition
MR imaging examinations were performed on a 3T Gyroscan Intera (Philips, Best, the Netherlands) with a transmit-receiver head coil. A 3D T1-weighted turbo-field-echo sequence was used for pituitary gland volumetry (matrix, 512 × 512; FOV, 256 × 256 mm2; TR, 7.4 ms; TE, 3.4 ms; 320 contiguous sagittal sections; section thickness, 0.5 mm; voxel size, 0.5 × 0.5 × 0.5 mm3, flip angle, 9°).
Pituitary gland volume was measured in sagittal sections by use of Multitracer (LONI Software, version 1.0; http://www.loni.ucla.edu/Software/MultiTracer)11 by a blinded, experienced radiologist (Fig 1A–B). Measurements were repeated in the same fashion after 4 to 6 weeks.
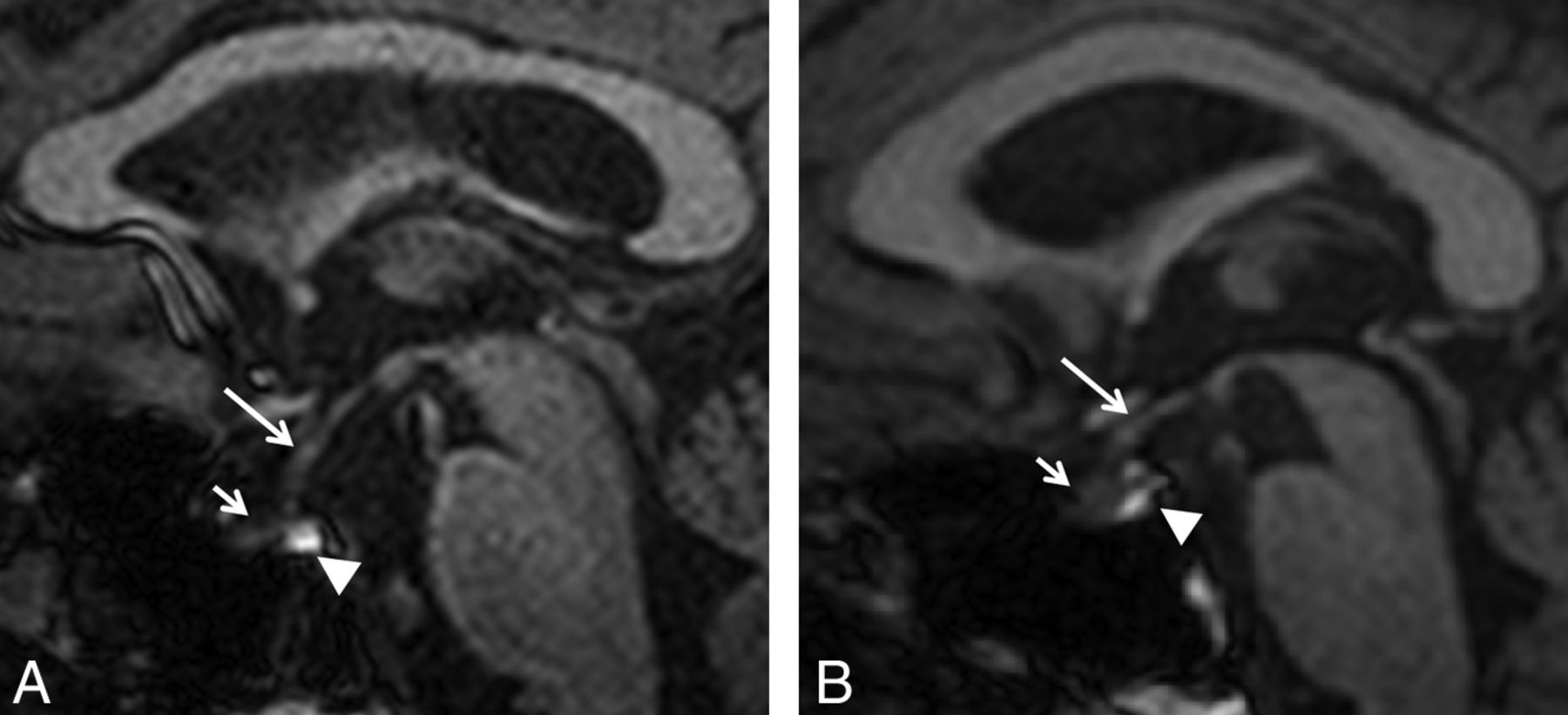
T1-weighted MR imaging shows midsagittal section of a normal subject, 57 years of age (A), and a patient of the same age with Kennedy disease (B). Infundibulum (long arrow), anterior lobe (short arrow), and posterior lobe (arrowhead) of the pituitary gland are shown.
Statistical Analysis
The volumetric data were analyzed by a univariant ANOVA with total brain volume as a covariant in a group comparison of patients and control subjects by use of SPSS 17.0 (IBM, Armonk, New York). Patient data, namely, pituitary volume, patient age, disease duration, and CAG repeat expansion length, were then also entered into a linear regression model.
Furthermore, a 2-tailed correlation analysis between pituitary gland volume and age of patients and control subjects, as well as a 1-tailed analysis for CAG repeat length in patients was performed.
Intraobserver reliability was evaluated with use of the Pearson correlation coefficient.
Results
Patients with KD had a mean pituitary volume of 636 (±90) mm3, whereas healthy control subjects had a volume of 534 (±91) mm3, respectively. Statistical analysis showed pituitary volume to be significantly higher in patients with KD than in healthy control subjects (P = .041). There was no significant difference in total brain volume (P = .379) (Table 1 and On-line Fig 1).
The correlation analysis showed that in healthy control subjects, pituitary gland volume was significantly lower in older individuals (Pearson correlation coefficient r = −0.712, P = .002), whereas patients with KD showed a trend toward increasing gland volume with age (r = 0.443) but without reaching statistical significance (P = .272) (On-line Fig 2).
Gland volume also correlated significantly with CAG repeat expansion length in the patient group (r = 0.630, P = .047) (On-line Fig 3).
Multiple linear regression showed that the CAG repeat expansion length was the only predictor of pituitary volume. Patient age and disease duration were excluded from the model and therefore were not predictive factors of pituitary volume in KD.
The overall Pearson correlation for intraobserver reliability was 0.94 (P < .001), which is generally considered to reflect an excellent reproducibility.
Discussion
In patients with KD, neurodegeneration has been attributed to a CAG repeat expansion in the AR. This mutation leads to intranuclear accumulation of AR, which can induce transcriptional dysfunction (toxic gain of function) causing cell death resulting in neurologic symptoms, whereas partial androgen insensitivity, on the other hand, appears to be caused by a loss of function from the same mutation in the first exon of the AR.12⇓–14 This loss of function leads to endocrine disturbances with symptoms such as gynecomastia, testicular atrophy with reduced fertility, diabetes mellitus, or hypercholesterolemia.1,15⇓–17 Our work is in line with previous reports on endocrinologic changes in patients with KD, with 6 of 8 patients presenting with gynecomastia, 1 patient presenting with erectile dysfunction, and 2 patients presenting with proven high testosterone levels. For instance, Dejager et al3 reported that >80% of cases presented with physical signs of androgen insensitivity, especially gynecomastia, undermasculinization, testicular hypotrophy, and reduced fertility, in a survey of 22 patients with KD. In most patients, gynecomastia was apparent before neurologic symptoms appeared; 50% showed additional signs of hypoandrogenicity such as erectile dysfunction and decreased sexual interest. As many as 86% exhibited a hormone profile indicative of partial androgen insensitivity. These changes slowly progressed over time.3 This evidence is corroborated by a large cohort of patients with KD in Japan in which Atsuta et al18 evaluated laboratory data in 61 cases. They stressed that even in older patients, testosterone levels remained high but decreased slightly with age.18
From the pathophysiologic point of view, testosterone normally inhibits LH and follicle-stimulating hormone (FSH) secretion through effects both on the hypothalamus and the pituitary gland.19 In patients with KD, partial androgen insensitivity frequently leads to high testosterone levels with elevated LH levels and hyper-responsiveness of LH to gonadotropin-releasing hormone as a possible sign of a larger stock of LH caused by a lack of suppressive effects of testosterone on the pituitary gland itself.3,20 Furthermore, the suppressive effect of synthetic androgens on testosterone levels, LH, and FSH is reduced in patients with KD, which suggests a weakened effect of androgens on the pituitary gland as a possible cause.21
LH and FSH are produced in the pituitary gland, which we investigated in the present study by means of high-field 3T MR imaging. The volume of the pituitary gland was significantly larger in patients with KD than in control subjects. It is known that pituitary gland size reflects hormonal secretion. An increase of LH and FSH at puberty, for example, leads to further enlargement.22,23 Volume also increases steadily with age in healthy subjects, in women up to a peak in the second decade and in men in the third decade.24 It could also be shown that high concentrations of gonadotropic hormones induced by the age-related drop in circulating gonadal steroids in climacteric women correlated with an increase of pituitary height. In men, pituitary height declines steadily after reaching the peak in the third decade.25
Although many studies could show that changes in pituitary dimensions predominantly affect the height of the gland,25 more recent studies also used a direct volumetric approach by use of 2D or 3D MR images.26,27
We therefore hypothesized that the lack of suppressive effects of testosterone on the pituitary gland in KD causing hypersecretion of LH may also lead to morphologic changes of the gland in KD.
Our analysis showed that patients with KD had a significantly higher pituitary volume as compared with normal, age-matched individuals, whereas there was no difference in total brain volume. This could reflect a hypertrophy of the pituitary gland as the result of elevated gonadotropic hormone secretion caused by the AR receptor mutation with partial androgen insensitivity.
Furthermore, patients with KD showed a trend to increasing gland volume in older patients, whereas control subjects showed a statistically significant decrease in volume. These findings are in line with previous studies on healthy individuals.24,25 These changes may reflect an ongoing process of gland hypertrophy over time and duration of the disease.
We also found larger pituitary gland volumes in patients with longer CAG repeat expansions. This is in line with the findings of Dejager et al,3 who described a significant association of the number of CAG repeats and endocrine changes, age of onset of gynecomastia, and laboratory findings.
In our data, CAG repeat expansion length also proved to be the only independent predictor of pituitary volume in patients with KD, whereas other factors such as patient age and disease duration were not predictive of pituitary volume. It seems likely, therefore, that the significant association of CAG repeat expansion length and clinical parameters found by Dejager et al3 is also reflected in the morphologic changes of the pituitary gland with even larger volumes in patients with longer CAG repeats. Patient age and disease duration alone were not sufficient to explain increasing gland volume.
There are limitations of the present study that must be observed. Laboratory findings concerning hormone levels at the time of the MR imaging were available in 2 patients with KD, showing elevated testosterone and SHBG levels. The endocrine changes described by Dejager et al3 must be expected in our collective of patients with genetically proved KD as well, especially because physical signs of androgen insensitivity were present in all cases. However, a connection between elevated hormone secretion and gland hypertrophy in KD could only be assumed, and further research with a full endocrine assessment is warranted.
Conclusions
In summary, we describe a significant increase of pituitary gland volume in patients with KD as compared with healthy individuals, previously not reported, as well as a positive correlation of gland size and CAG repeat expansion length. This could reflect hypertrophy of the pituitary gland as the result of elevated gonadotropic hormone secretion caused by the CAG repeat expansion in the AR with partial androgen insensitivity.
REFERENCES
- Received December 31, 2012.
- Accepted after revision February 25, 2013.
- © 2013 by American Journal of Neuroradiology





{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.