Abstract
SUMMARY: Bosch-Boonstra-Schaaf optic atrophy syndrome (BBSOAS) is a rare autosomal dominant syndrome secondary to mutations in NR2F1 (COUP-TF1), characterized by visual impairment secondary to optic nerve hypoplasia and/or atrophy, developmental and cognitive delay, and seizures. This study reports common neuroimaging findings in a cohort of 21 individuals with BBSOAS that collectively suggest the diagnosis. These include mesial temporal dysgyria, perisylvian dysgyria, posterior predominant white matter volume loss, callosal abnormalities, lacrimal gland abnormalities, and optic nerve volume loss.
ABBREVIATIONS:
- BBSOAS
- Bosch-Boonstra-Schaaf optic atrophy syndrome
- COUP-TFI
- chicken ovalbumin upstream promoter transcription factor 1
Bosch-Boonstra-Schaaf optic atrophy syndrome (BBSOAS, Online Mendelian Inheritance in Man 615722) is a rare autosomal dominant disorder primarily characterized by visual impairment from optic hypoplasia and/or atrophy and neurocognitive and developmental delay.1 A wide variety of additional clinical phenotypes has been reported in the literature in subsets of patients, including hypotonia, oromotor dysfunction, hearing abnormalities, seizures, autism spectrum disorder, and personality disorders such as attention deficit/hyperactivity disorder and obsessive compulsive disorder.2
BBSOAS is caused by mutations, nearly all de novo, in the nuclear receptor subfamily 2 group F member 1 (NR2F1) gene responsible for encoding the NR2F1 protein, which is also known as the chicken ovalbumin upstream promoter transcription factor 1 (COUP-TFI). This protein stimulates initiation and regulation of transcription but also serves as a nuclear hormone receptor. NR2F1 is important for myriad central nervous embryologic developments that include ocular globe and optic nerve development as well as cortical development, axonal guidance, neurogenesis and neuronal arborization, hippocampal volume, and functional organization.3⇓⇓⇓⇓⇓-9
In this report, we describe readily identifiable, common neuroimaging findings of BBSOAS that strongly suggest this genetic diagnosis.
CASE SERIES
Case Selection
A clinical database of multi-institutional (18 total) patients genetically diagnosed with BBSOAS was queried after institutional review board approval at Baylor College of Medicine. This database included patients seen at a single institution during the historic discovery of this unique genetic entity. A total of 51 individuals with BBSOAS were documented at a single institution, having undergone genetic and clinical evaluation. Neurologic and ophthalmologic information was recorded. Brain MR imaging examinations of patients were centrally collected and were available for 21 patients.
Imaging Evaluation
All imaging was performed on 1.5T or 3T MR imaging units. All studies included standard departmental multiplanar T1-weighted, T2-weighted, FLAIR, and DWI sequences of the brain and orbits with expected variations in the protocols per the multiple institutions. Most of the studies were performed without contrast. In a small number of patients in whom intravenous contrast was administered, the T1-weighted postcontrast imaging was reviewed but was noncontributory in all cases and will not be discussed further. Brain MR imaging examinations of all patients were collectively consensus-reviewed by 2 board-certified pediatric neuroradiologists (N.K.D. and S.F.K.), each with 10 years of experience. Neuroimaging findings of the orbit, optic nerve apparatus, cortical gray and hemispheric white matter of the cerebrum, cerebellum, vermis, central gray matter, and midline structures were categorically reviewed and recorded. For the corpus callosum, qualitative and quantitative evaluations were performed. Quantification included measurement of the overall callosal anterior-posterior dimension (also called callosal length) as well as segmental thickness of the genu, body, isthmus, and splenium. Callosal measurements of ≤ 3rd percentile or ≥ 97th percentile were deemed abnormal compared with the previously published reference measurements by Garel et al.10 Pertinent data-based medical history was retrospectively reviewed, including demographics, clinical signs and symptoms, as well as the results of a detailed neuro-ophthalmologic evaluation.
RESULTS
Demographic and Clinical Findings
The mean age of the children at MR imaging was 4.5 years (range, 0.33–16 years) with 12/21 (57%) being female. A variety of de novo pathogenic variants in NR2F1 were present in this cohort, most commonly missense mutations in NR2F1, present in 9/21 (43%) patients (Table 1).
Pathogenic variants in NR2F1
Ophthalmologic abnormalities are a hallmark of BBSOAS with general visual impairment present in 19/21 (90%) patients of this cohort, of which many (1 patient without specific documentation) were found to have cerebral visual impairment (10/20, 50%). A cerebral visual impairment diagnosis was based on clinical examination by 2 pediatric neuro-ophthalmologists of visual behavior not consistent with anterior visual pathway dysfunction (ie, optic nerve abnormality) alone and clinically observed features (vision inattention, low-light gazing, difficulty with visual complexity) consistent with a cerebral visual impairment diagnosis. Furthermore, a cerebral visual impairment diagnostic screening questionnaire was given to all patients’ families. Optic nerve atrophy, optic nerve hypoplasia, or both were noted in 18/21 (86%) and 8/20 (40%) patients, respectively (1 patient did not have specific documentation on the presence or absence of optic nerve hypoplasia). Nystagmus and alacrima (decreased tear production) were seen in 12/20 (60%) (1 patient without specific documentation) and 15/19 (79%) patients (2 patients without specific documentation), respectively (Table 2). Detailed ophthalmologic findings will be discussed in a forthcoming publication.
Ophthalmologic phenotypes
Developmental data were available for 19/21 (90%) patients. Intellectual, and speech delay were common in the group, seen in 16/19 (84%) and 17/19 (89%) patients. Autistic features were present in 16/19 (84%) patients. Most interesting, despite such delays, 14/17 (82%) patients were clinically reported to have unusually strong long-term memory ability (2 patients without specific documentation). Motor impairment including general motor delay, oromotor dysfunction, and hypotonia were similarly present with high frequency in 15/19 (79%), 16/19 (84%), and 18/19 (95%) patients, respectively. Seizures or infantile spasms were present in 12/19 (63%) patients (Table 3). Detailed genotypic-phenotypic findings have been previously published by this group.11
Neurocognitive phenotypes
Ophthalmologic Imaging
Eighteen of 21 patients (86%) demonstrated bilateral symmetric volume loss consistent with optic atrophy and/or hypoplasia of the optic apparatus, including of the optic nerves, chiasm, and optic tracts (Fig 1). Such volume loss was subjectively noted to be of variable severity from mild to moderate to severe. These findings correlated with the presence or absence of optic nerve hypoplasia and/or atrophy in all patients. Of the 3/21 (14%) patients with normal optic nerves on imaging, 1/3 patients (67%) had abnormal ophthalmologic examination findings with atrophy. Data on optic nerve hypoplasia were not available in 1/21 (5%) patients.

Coronal T2WI in multiple patients with BBSOAS. Bilateral, symmetric, severe optic nerve volume loss in patient 19, a 6-year-old boy (A) (black arrows); mild-to-moderate volume loss in patient 18, a 3-year-old girl (B) (black arrows); and normal optic nerves in patient 11, a 4.7-year-old girl (C) (black arrows).
An interesting clinical hallmark reported in 15/19 (79%) patients in this cohort is alacrima or decreased tear production. Thirteen of 21 (62%) patients had hypoplastic bilateral lacrimal glands on MR imaging, and 8/21 (38%) patients had normal lacrimal glands. Data on tear production were not available in 2/21 (10%) patients. One of 19 (5%) patients with hypoplastic lacrimal glands did not have abnormal tear production. Three of 19 (16%) patients with normal lacrimal glands on imaging had normal tear production. Five of 19 (26%) patients with normal lacrimal glands on imaging had decreased tear production. Ten of 19 (58%) patients with hypoplastic lacrimal glands had absent or decreased tear production (Fig 2).

Patient 2, an 0.8-year-old girl. Coronal T1WI of the orbits demonstrates hypoplastic lacrimal glands (arrows) (A). Patient 11, a 4.7-year-old girl. Coronal T2WI image of the orbits demonstrates normal lacrimal glands (arrows) in comparison (B).
Midline Imaging
Fourteen of 21 (67%) patients had an abnormal corpus callosum (Fig 3). We observed several different imaging phenotypes:
Thickened genu and body, thinned splenium with normal callosal length (1/21) (5%)
Thickened body and normal callosal length (1/21) (5%)
Thickened body and splenium with normal callosal length (1/21) (5%)
Thickened genu and body and normal callosal length (1/21) (5%)
Thinned splenium with normal callosal length (3/21) (14%)
Thinned body with normal callosal length (1/21) (5%)
Thinned splenium with decreased callosal length (2/21) (10%)
Thinned body, isthmus, and splenium with decreased callosal length (1/21) (5%)
Normal thickness with isolated decreased callosal length (1/21) (5%)
Thickened genu and body and thinned splenium with decreased callosal length (1/21) (5%)
Thickened body and thinned splenium with decreased callosal length (1/21) (5%)
Normal corpus callosum (7/21) (33%)
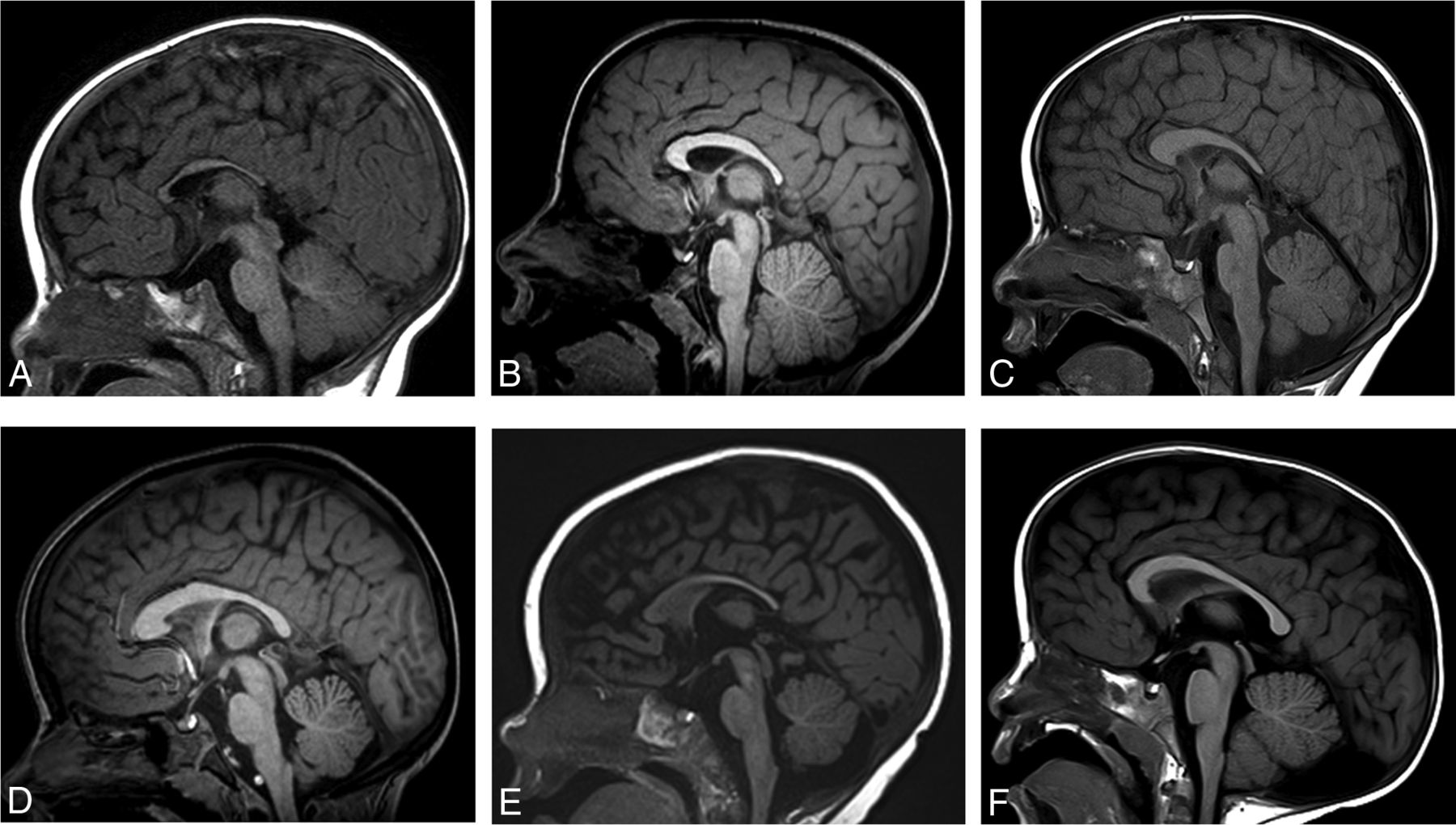
Midline sagittal T1WI in multiple patients with BBSOAS. Patient 5, a 0.5-year-old boy. Decreased callosal length with a thinned body, isthmus, and splenium of the corpus callosum (A). Patient 20, a 5-year-old boy. Decreased callosal length with thinning of the isthmus (B). Patient 8, a 2-year-old boy. Decreased callosal length with a thickened genu and body but a thinned splenium (C). Patient 21, a 6-year-old girl. Normal callosal length, thickened genu and body with a thinned splenium (D). Patient 14, a 0.6-year-old girl. Normal callosal length with a thinned splenium (E). Patient 9, a 6-year-old boy. Normal corpus callosum. Incidental note is made of a retrocerebellar arachnoid cyst (F).
All patients had a normal septum pellucidum and hypothalamic pituitary axis. One of 21 (5%) patients had a hypoplastic left olfactory bulb. One of 21 (5%) patients demonstrated subjective decreased volume of the pons. A separate patient, 1/21 (5%), demonstrated a vermian cleft. One of 21 (5%) patients, an 8-month-old infant (patient 15), demonstrated T2 hyperintensity of the bilateral central tegmental tracts. Otherwise, midline posterior fossa anatomy, including the brainstem and vermis, was normal in all patients.
Lobar Imaging
Twenty-one of 21 (100%) patients had dysgyria, defined as an abnormal gyral pattern with abnormal sulcal depth or orientation but with normal cortical thickness and normal gray-white matter differentiation along the bilateral mesial temporal lobes. Perisylvian dysgyria was present in 15/21 (71%) patients, occurring bilaterally in 14/15 (93%) patients. Perisylvian dysgyria in our cohort group included a variety of different radiologic phenotypes. These principally included ≥1 of the following: craniocaudal elongation of the temporal lobes, lateral temporal dysgyria, and posterior perisylvian dysgyria involving the inferior parietal lobule with posterior elongation of the Sylvian fissure or other generally dysmorphic Sylvian fissures including, for example, underopercularization and steep craniocaudal angulation of the Sylvian fissure axis (Figs 4–6). There were no abnormalities of myelination to suggest leukodystrophy. None of the patients had gray matter heterotopia or cortical dysplasia.

Coronal 3D T1WI (A–C) demonstrating normal anatomy of the temporal lobes as a reference. PHG indicates parahippocampal gyrus; CS, collateral sulcus; OTG, occipitotemporal gyrus; OTS, occipitotemporal sulcus; ITG, inferior temporal gyrus; ITS, inferior temporal sulcus; MTG, middle temporal gyrus; STS, superior temporal sulcus; STG, superior temporal gyrus; SF, Sylvian fissure.

Patient 6, a 16-year-old boy. Coronal 3D T1WI demonstrates dysmorphic Sylvian fissures (stars, A). The anterior temporal lobes appear large with signficant dysgyria (A) and craniocaudal elongated morphology. Dysgyria is noted in the bilateral mesial temporal lobes (boxes, B). Dysgyria broadly involves the posterior right lateral temporal lobe, appearing as gross overgyration with small gyri and shallow sulci (C), with right mesial temporal lobe dygyria still noted. The right Sylvian fissure is signficantly asymmetric to the left (arrows), with an exaggerated upslope (C).

Patient 21, a 6-year-old girl. Axial T2WI demonstrates dysgyria of the mesial temporal lobes (stars, A). Coronal T2WI demonstrates dysgyria of the mesial temporal lobes (box, B). Dysgyria broadly involves the posterior right lateral temporal lobe. Both temporal lobes appear elongated craniocaudally. The right Sylvian fissure is signficantly asymmetric to the left (arrows), with an exaggerated upslope (B). Bilateral perisylvian dysgyria is present. Normal findings on axial and coronal T2WI are shown for comparison (C–D). An enlarged perivascular space is incidentally noted on the right (arrow, C). Note the normal, mostly horizontal axis of the Sylvian fissures and the normal anatomy of the temporal lobes and perislyvian parenchyma (arrows) (D).
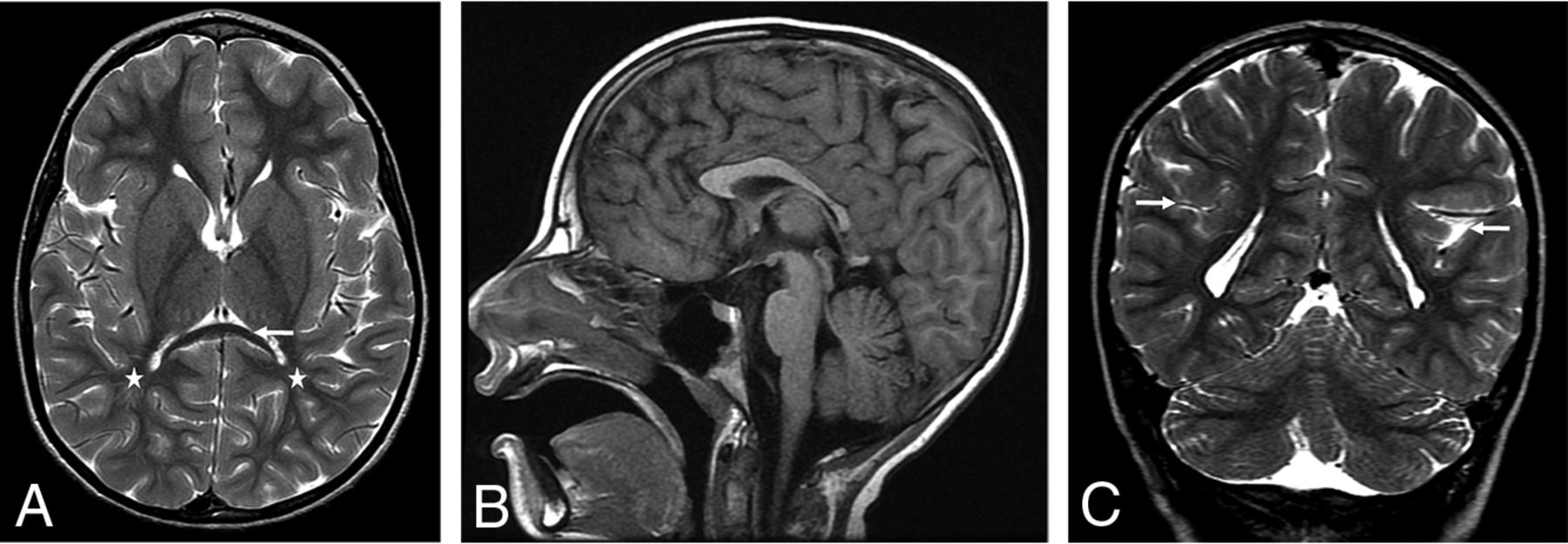
Decreased cerebral white matter volume was present posteriorly in 11/21 (52%) patients. Decreased anterior and posterior cerebral white matter volume was present in 3/21 (14%) patients (Fig 7). Twelve of 14 (86%) patients with lobar white matter volume loss had abnormalities of the corpus callosum. Of the 18/21(86%) patients with optic nerve abnormalities on imaging, 12/18 (67%) demonstrated lobar white matter volume loss.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patient 1, an 8 year-old girl. Axial T2WI demonstrates signficant posterior white matter volume loss (stars, A). Note the thinning of the splenium of the corpus callosum (arrow), similarly seen on the sagittal T1 image (B). Coronal T2WI demonstrates dysmorphic Sylvian fissures bilaterally, with an exaggerated upslope of the fissures bilaterally. Bilateral perisylvian dysgyria is present with craniocaudal elongated temporal lobes bilaterally (arrows, C).
One of 21 (5%) patients had a cavernous malformation.
DISCUSSION
BBSOAS is a rare autosomal dominant disorder due to loss-of-function mutations in NR2F1, also known as COUP-TF1, originally described in 2014 by Bosch et al.1 Patients are characterized by visual and neurocognitive deficits but may also manifest additional clinical symptoms of hypotonia, seizures, autism spectrum disorders, oromotor dysfunction, and hearing abnormalities.
Neuroimaging findings in BBSOAS have been partially reported in the literature to date. Thinning of the corpus callosum has been reported by multiple authors, including 8/15 patients by Chen et al.2,12 We similarly discovered abnormalities in the corpus callosum in 67% patients. However, callosal abnormalities were quite heterogeneous with variable combinations of the following findings: normal or decreased anterior-posterior callosal length and segmental thinning and/or thickening. All patients in this study had a normal septum pellucidum and a normal hypothalamic pituitary axis.
Thinning of the optic apparatus has also been expectedly reported in previous literature, corresponding with ophthalmologic findings common to these patients.2,13,14 Nearly all of the patients in our group (86%) demonstrated bilateral optic nerve volume loss of variable severity with all other patients demonstrating normal optic nerves. Additional orbital findings in our patient population included subjectively hypoplastic (13/21) lacrimal glands, which, in a subset of patients, did correlate with alacrima.
As noted, NR2F1 is important for myriad central nervous embryologic developments that include cortical development, axonal guidance, neurogenesis and neuronal arborization, and hippocampal volume and functional organization.3⇓⇓⇓⇓⇓-9 Recently, Bertacchi et al12 reported altered gyration, described as being polymicrogyria-like in the supramarginal and angular gyrus of only the left inferior parietal lobule. These authors also reported elongation of the right superior occipital gyrus and prominent occipital gyration in a separate patient. We, similarly, found abnormal gyration and sulcation of the perisylvian parenchyma in 15/21 (71%) patients, of which 14/15 (93%) were bilateral, but we also report more common similar findings in the mesial temporal lobes bilaterally in 21/21 (100%) patients. These imaging findings are best termed dysgyria, defined as an abnormal gyral pattern with abnormal sulcal depth or orientation but with normal cortical thickness and normal gray-white matter differentiation.
While Bertacchi et al12 described this anomalous gyration as polymicrogyria-like, the findings are not polymicrogyria (as they note), given the lack of cortical thickening as evidenced by the sample images in their publication. None of the patients in our cohort had findings of cortical dysplasia or gray matter heterotopia, and this has not been reported elsewhere, to our knowledge. As Bertacchi et al noted, much work is needed to better understand the role of NR2F1 in human cortical folding patterns, gyri and sulci positioning, and the effect of NR2F1 on progenitor subtypes that ultimately lead to the neuroradiologic phenotypes discussed here and their eventual clinical phenotypic expressions.12
Mesial temporal dysgyria is not specific to BBSOAS. In fact, it has been well-reported with other, unrelated entities, including, for example, Apert syndrome due to mutations in FGFR2. Similarly, achondroplasia, thanatophoric dysplasia, and hypochondroplasia, all of which are due to mutations in FGFR3, are typified by such dysgyria.15,16 Furthermore, perisylvian dysgyria, an entity seldom reported in the literature, is not specific to BBSOAS, having recently been described in individuals with the ACTA2 gene mutation.17 Dysgyria, in general, has also been described in patients with microtubule mutations.18 Thematic to dysgyria in these examples, however, is the linkage of mutations in genes important for axonal guidance and neuronal migration.
Many of our patients (14/21) demonstrated subjective posterior or anterior and posterior cerebral white matter volume loss, with 12/14 (86%) patients with lobar white matter volume loss having abnormalities of the corpus callosum. Similarly, of the 18/21 (86%) patients with optic nerve abnormalities on imaging, 12/18 (67%) demonstrated lobar white matter volume loss.
A limitation of our study is the heterogeneous imaging performed in the cohort, because patients and therefore imaging were collected from a variety of institutions. Most imaging performed was, therefore, nonvolumetric conventional 2D imaging, thwarting our ability to perform systematic volumetric analysis of the whole and ultrastructural brain. Given our findings and those already in the literature, volumetric analysis would be of special interest at the level of the temporal lobes and the limbic system. Most interesting, unusually strong long-term memory was found in 82% of patients, despite most demonstrating intellectual disability, indicating that additional quantification of brain volumetry may provide insights into this disorder. Similarly, advanced imaging including DTI or functional MR imaging was not uniformly available to evaluate the microstructural white matter architecture or connectivity of the brain. Such analyses may prove revealing in the deeper understanding of NR2F1 gene function as it relates to the human brain. In Nr2f1+/− mice, MR imaging has already demonstrated decreased hippocampal volumes and increased volume of the caudate nucleus, putamen, and neocortex, with preserved neuronal cell density foreshadowing the potential of these MR imaging techniques.19
CONCLUSIONS
Patients with BBSOAS commonly manifest abnormalities of the optic pathway, lacrimal glands, corpus callosum, and dysgyria of the temporal lobes and perisylvian cortex, which, in combination, can suggest this disorder and indicate a need for genetic testing.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- Received June 20, 2022.
- Accepted after revision December 6, 2022.
- © 2023 by American Journal of Neuroradiology