
Abstract
Summary: We report three patients in whom neurologic symptoms and cortical laminar necrosis developed after immunosuppressive treatment (cyclosporin A and FK 506) and polychemotherapy (vincristine and methotrexate). Initial neuroradiologic studies showed cortical and white matter involvement. Follow-up studies showed cortical hyperintense lesions on T1-weighted MR images, consistent with cortical laminar necrosis. The clinical and radiologic data indicate that a transient hypoxic-ischemic process could have been responsible for the encephalic lesions in these three patients.
Acute neurologic effects produced by immunosuppressive agents have been widely described. The frequency of cyclosporin A (CsA)-induced neurotoxicity is thought to be between 25% and 35% (1, 2), and FK 506 neurotoxicity has been found in 32% of patients undergoing orthotopic liver transplantation (3). Vincristine-induced neurotoxicity is also well documented, but only a few cases of cortical blindness have been reported (4, 5).
Several theories have been proposed to explain the exact mechanism through which these agents cause neurotoxicity. One hypothesis is related to hypertensive encephalopathy and vascular injury. In this article, we report three patients treated with immunosuppressive drugs and chemotherapy (vincristine, methotrexate) in whom cortical laminar necrosis developed. The neuroradiologic findings suggest a transient hypoxic pathogenesis.
Case Reports
Case 1
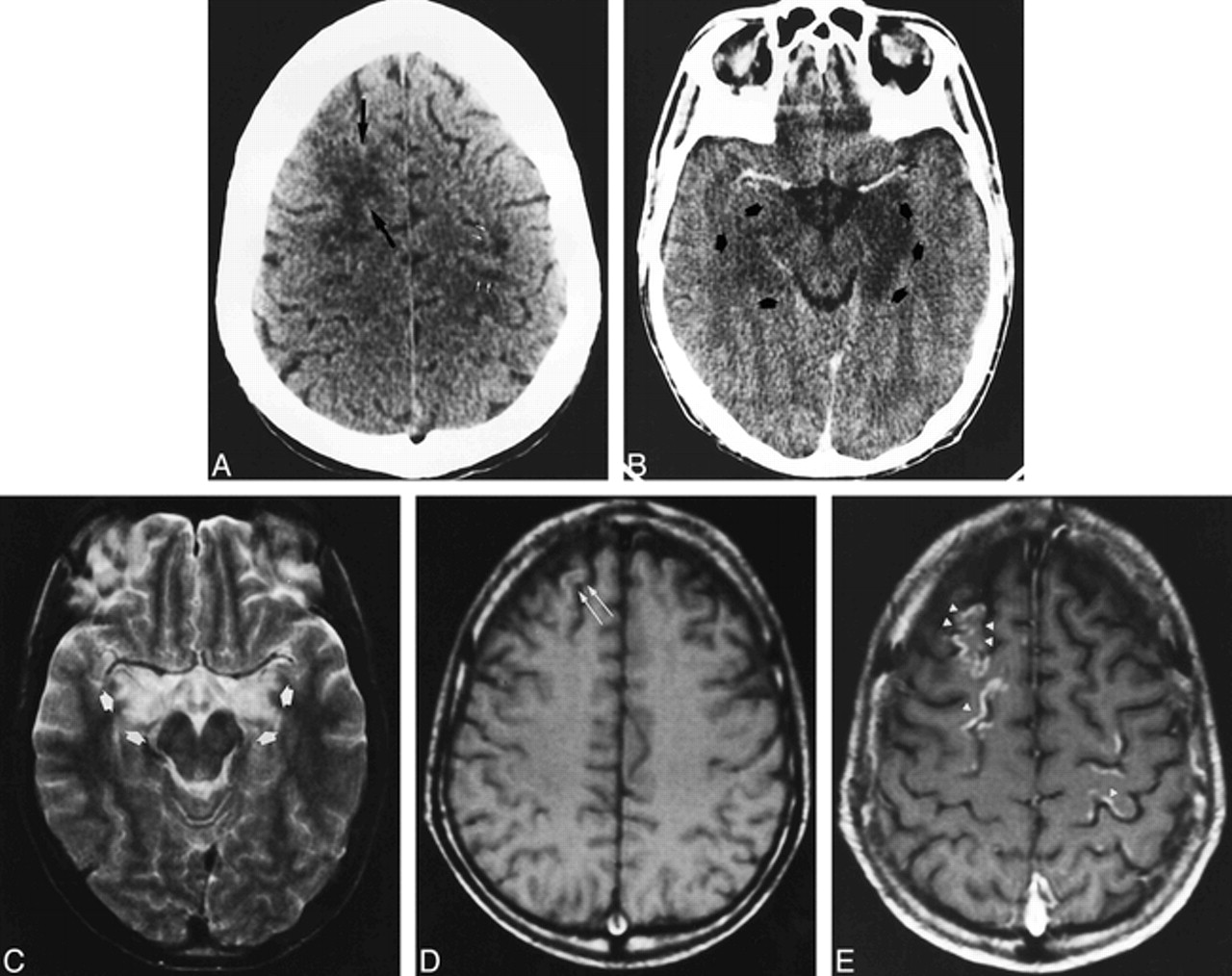
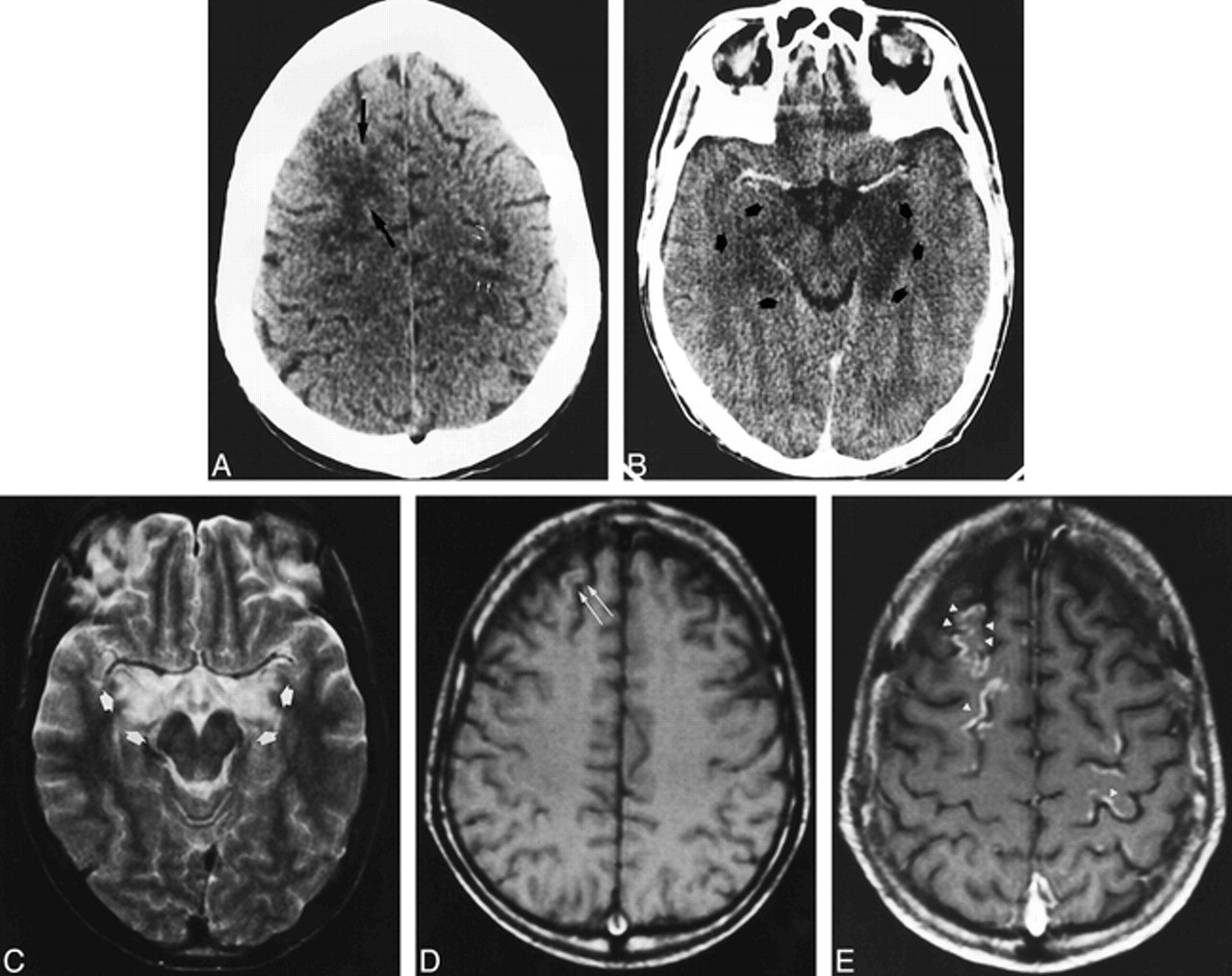
A 54-year-old woman with chronic renal failure underwent renal transplantation without surgical complications. Initial immunosuppressive therapy consisted of FK 506 and steroids. Thirty-five days after treatment the patient experienced emotional lability, which was assumed to be a depressive syndrome. A few days later, temporospatial disorientation and stupor developed. The initial neurologic examination revealed eye deviation to the left, bilateral vertical-rotator nystagmus, and left hemiparesis. Although all these symptoms resolved without treatment, 12 hours later the patient exhibited eye deviation to the right with horizontal nystagmus and right hemiparesis, but right Babinski response was not present. Partial seizure in the right arm, tonic-clonic generalized seizure, altered consciousness, and coma developed progressively. FK 506 blood levels at this time were 14 ng/mL (mean values, 5–15 ng/mL). Laboratory studies revealed hypoalbuminemia without other abnormalities. Administration of FK 506 was canceled, and 48 hours later the neurologic signs resolved by themselves and consciousness returned to normal. Initial CT examination showed subcortical white matter occipitoparietal hypodensities with cortical involvement in the posterior parietal lobes (Fig 1A) and involvement of the right cerebellum. An MR imaging study performed the same day showed extensive involvement of the cortex and white matter in the frontal, parietal, temporal, and occipital lobes. Involvement of the right thalamus, both caudate nuclei, the left lenticular nuclei, both internal capsules, the right mesencephalon, the pons, and the cerebellum was also observed (Fig 1B and C). Enhancement in the cerebral cortex with a gyral pattern after administration of contrast material was noted. A second MR imaging study performed after the resolution of the neurologic clinical signs showed improvement of the lesions in both the occipitoparietal and frontal lobes and the cerebellum.

Case 1: 54-year-old woman who underwent renal transplantation and immunosuppressive therapy with FK 506.
A, Contrast-enhanced axial CT scan obtained at the onset of symptoms shows subcortical bilateral parietal hypodensities (arrows).
B and C, Axial T2-weighted MR images 12 hours later show diffuse cortical and white matter hyperintensities involving the bilateral frontal and parietal lobes (B). Involvement of right thalamus (black arrow), white matter, and both occipital lobes is also observed (white arrows) (C).
D and E, Follow-up MR studies 3 weeks later show complete resolution of the initial lesions. Axial T2-weighted image at the same position as B (D) shows no abnormalities. Axial T1-weighted unenhanced image (E) shows curvilinear areas of hyperintensity of superficial cortical layers in the parietal region (arrows).
Another MR examination, 3 weeks later, showed complete resolution of the initial lesions (Fig 1D), but T1-weighted images revealed the appearance of gyral hyperintense lesions in the frontal, parietal, and occipital cortex (Fig 1E).
Case 2
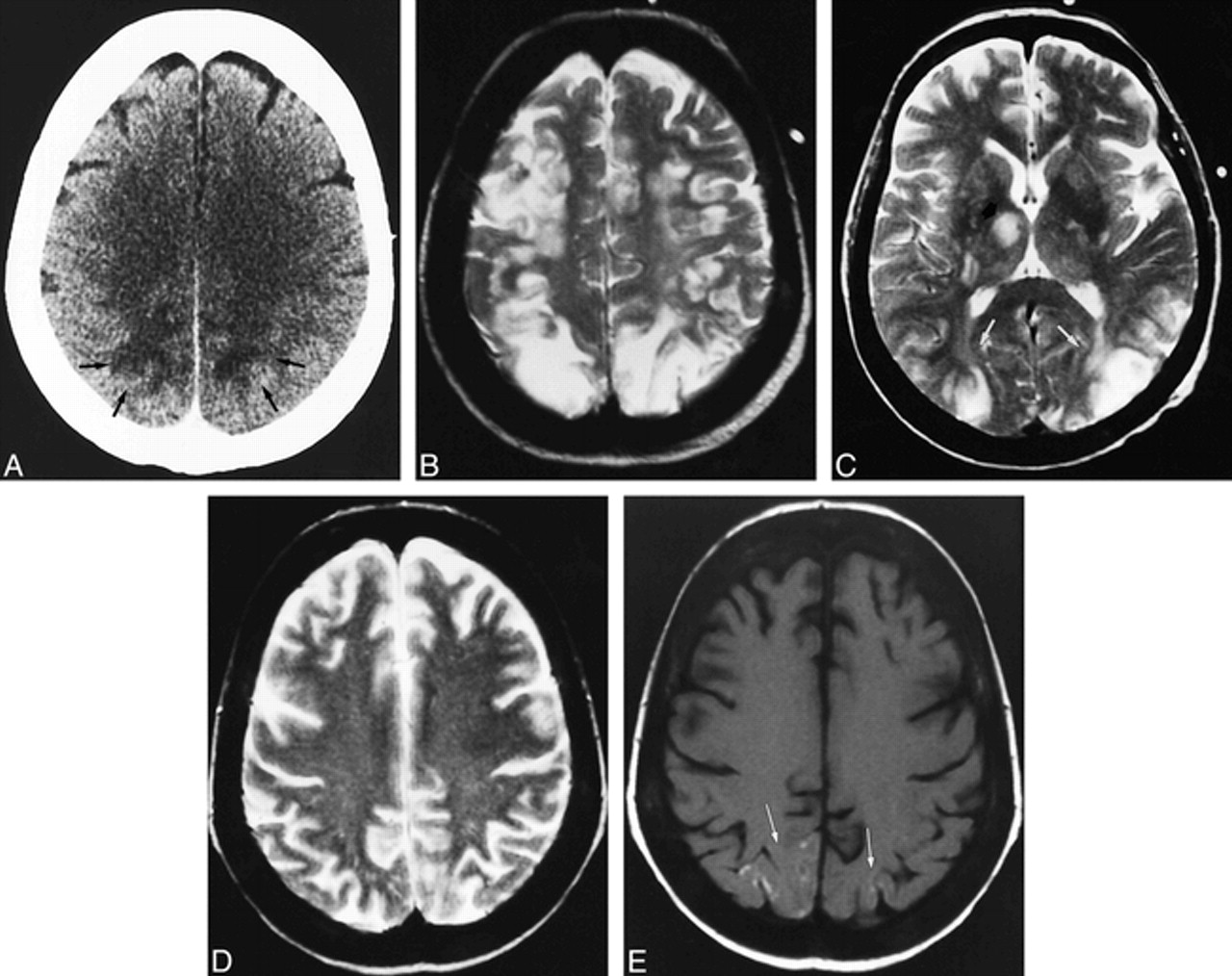
A 28-year-old man with acute myeloid leukemia (LAM 5b) was treated with polychemotherapy and an autogenic bone marrow transplantation. He remained in complete remission until 17 months later, when he suffered a relapse and was admitted to our hospital for an allogenic bone marrow transplantation after undergoing a new polychemotherapeutic course. Immunosuppressive treatment included CsA (40–90 mg/12 h) in oral doses. On day 7 after transplantation, a cutaneous rash developed in association with graft-vs-host disease, and the dosage of CsA was increased to 125 mg/12 h. On day 19, the patient appeared apathetic, which was initially assumed to be a depressive syndrome. Progress was unfavorable, with onset of decreased consciousness, temporospatial disorientation, akinetic mutism, and, finally, right hemiparesis. Blood CsA levels were 238 ng/mL (normal range, 100–400 ng/mL). Biochemical data revealed only hypoalbuminemia and hyponatremia (minimal blood levels were 120 mEq/L), which was treated and corrected. The patient partially improved, but loss of short-term memory persisted. CT findings were normal. On day 30 after the transplantation, mutism, mydriatic pupils, and right arm paresis developed. Deep tendon reflexes were hyperactive in the right extremities. There was no high blood pressure, and biochemical parameters were normal. The dosage of CsA administered was 175 mg/12 h, and the blood level of CsA was 160 mg/mL. The neurologic symptoms gradually resolved by themselves over the following days, although CsA doses were maintained. A new CT examination with contrast material showed cortical and white matter hypodensities involving the frontal and parietal lobes (Fig 2A) as well as the hippocampal cortex (Fig 2B). A CT scan obtained 6 days after the onset of symptoms showed partial improvement of the lesions. An MR examination 20 days later revealed slightly hyperintense lesions on T2-weighted images in the frontal and parietal lobes, and a decrease of hippocampal involvement (Fig 2C). Slight hyperintensities of the superficial cortical layers were visible on T1-weighted images (Fig 2D). Contrast-enhanced T1-weighted images showed cortical enhancement with gyral distribution in the frontoparietal lobes, with no abnormalities in the adjacent white matter (Fig 2E).
Case 2: 28-year-old man who underwent bone marrow transplantation and immunosuppressive therapy with CsA.
A and B, Axial CT scans obtained at the onset of right arm paresis, mutism, and mydriatic pupils show confluent hypodensity in the right frontal white matter (black arrows) and patchy areas of hypodensity in the left prerolandic area (white arrows) (A). After contrast injection (B), there is marked hypodensity of the left hippocampus and slight hypodensity in the right hippocampus (arrows).
C–E, MR studies 20 days later. Axial T2-weighted image at the level of the midbrain (C) shows partial resolution of the left hippocampal lesion with remaining bilateral hippocampal hyperintensities (arrows). Axial noncontrast T1-weighted image in the frontoparietal region (D) shows a subtle hyperintensity in the right frontal cortex (arrows). T1-weighted image after contrast administration (E) shows gyral enhancement in both frontal and parietal lobes (arrowheads).
Case 3
An 18-year-old woman was hospitalized for a check-up and for treatment of acute lymphoid T leukemia. She initially underwent polychemotherapy in accordance with a protocol that consisted of vincristine, methotrexate, daunorubicin, corticoids, intrathecal methotrexate, and prednisone. She reported a headache from the first day of hospitalization. Laboratory evaluation revealed high levels of LDH and creatinine, which were assumed to be due to tumoral lysis, and hypercalcemia, attributed to paraneoplasic syndrome. These parameters returned to normal on the following days. Findings on a brain CT scan were normal. On day 9 after polychemotherapy, the patient experienced visual disturbances and generalized tonic-clonic seizure; 24 hours later, she suffered two partial seizures, cortical blindness, and right hemiparesis. CSF showed a protein level of 100 mg/dL, 20 cells/mm3, and 90% segmented neutrophils. There was no high blood pressure, and blood levels of cholesterol and magnesium were normal. The patient was treated with anticonvulsant therapy and antibiotics; the polychemotherapy protocol remained unchanged. Two days later, these symptoms had almost disappeared, and only a slight nystagmus was observed. A subsequent brain CT scan revealed nonenhancing areas of hypoattenuation in the parietal, frontal (Fig 3A), and occipital white matter. An MR examination the same day showed diffuse cortical and white matter involvement of the cerebral lobes, cerebellum, and right hemimesencephalon (Fig 3B and C). A follow-up MR imaging study, 3 days later, showed nearly complete resolution of the abnormalities (Fig 3D), and only cortical hyperintense lesions with gyral distribution in the occipital lobe and both frontoparietal lobes remained on T1-weighted images (Fig 3E).

Case 3: 18-year-old woman treated for acute lymphoid T leukemia with polychemotherapy, including vincristine and methotrexate.
A, Axial contrast-enhanced CT scan obtained at the onset of neurologic symptoms shows right parietal white matter hypodensity (black arrows) and subtle patchy hypodensities in the frontal white matter (white arrows).
B and C, MR study performed the same day. Axial T2-weighted image in the posterior fossa (B) shows diffuse bilateral cerebellar lesions. Axial T2-weighted image in the frontoparietal lobes (C) shows thickened cortex of both frontal and parietal lobes with parietal white matter involvement.
D and E, MR study 11 days later. T2-weighted image in the frontoparietal region (D) shows complete resolution of the initial lesions. Corresponding axial noncontrast T1-weighted image (E) shows laminar hyperintensities in the bilateral frontal cortex (arrows).
Discussion
A wide variety of neurologic side effects may occur with the use of FK 506, which are similar to those associated with CsA. These effects have been classified as major and minor, depending on the nature of the clinical impact rather than on eventual outcome. The minor effects include insomnia, visual symptoms, headache, tremors, and mood changes; they are more frequent than major effects, occurring in 40% to 60% of transplant patients (3, 6, 7). The major side effects include akinetic mutism, encephalopathy, seizures, and focal neurologic disturbances; they occur in 5% to 8% of transplant recipients.
Neuroimaging abnormalities related to FK 506 have been less frequently reported in the literature, and findings on MR and CT studies are usually similar to those observed in CsA-induced neurotoxicity (7–9). The most common changes seen in CsA-induced neurotoxicity on imaging studies are nonenhancing areas of hypoattenuation on CT scans and T2 prolongation in the parietal and occipital white matter on MR images. These changes are usually reversible. Jansen et al (1) reported one patient treated with CsA who had cortical T1-weighted hyperintensities, such as are found in cortical laminar necrosis. Appignani et al (7) described the neuroimaging findings in six patients with FK 506-neurotoxicity. These patients had reversible lesions in the parietal, occipital, and frontal white matter, and also in the pons, basal ganglia, and cerebellum.
The precise mechanism of FK 506-related neurotoxicity remains unknown, and diverse theories have been proposed, which are similar to those associated with neurotoxicity-induced by CsA. In the early studies, it was hypothesized that metabolic disturbances, such as hypocholesterolemia and hypomagnesemia, produced by CsA and FK 506 could be the cause of neurotoxicity (10). But this theory has not been confirmed in recent reports.
One of the most postulated hypotheses is related to hypertensive encephalopathy, which corresponds to the white matter changes observed principally in the parietal and occipital lobes seen on CT and MR studies (11). Theories regarding the pathophysiology of CsA- and FK 506-induced hypertension remain controversial, and the majority of the patients reported in the literature had no history of high blood pressure at clinical onset. None of the three patients in our series had ever had a hypertensive episode.
Truwit et al (2) suggested that CsA-induced neurotoxicity may be related to endothelin, a neuropeptide released by endothelial cells that acts on vascular smooth cells, resulting in an intense vasoconstriction. Specific binding sites for endothelin have been identified in several areas of the human brain, including the cerebellum, hippocampal formation, diencephalon, and choroid plexus. In the kidneys, CsA and FK 506 have been shown to disrupt endothelial integrity, which includes an increased release of endothelin (12). If endothelial integrity were disrupted, these drugs could gain access to astrocytes. Endothelin also could gain access to the cerebral vascular smooth muscle, resulting in vasoconstriction and vasospasm. Elevated circulating endothelin could promote systemic hypertension. Under such conditions, local ischemia and consequent white matter edema would be likely. If systemic hypertension also occurred, some of the neuroradiologic findings described in the literature, such as neurotoxicity, could show typical transient white matter edema in the subcortical parietal and occipital lobes, as observed in cases of acute hypertensive encephalopathy. If the vasospasm is prolonged, a hypoxic episode could result, and lesion location would be similar to that described in hypoxic encephalopathy (13).
In our two patients treated with immunosuppressive agents, the evolution of the findings on the imaging studies indicates that they probably suffered a hypoxic episode. Initially diffuse cortical and white matter involvement, and lesions in the basal ganglia, pons, and cerebellum, indicated cytotoxic and vasogenic edema (Figs 1A-C and 2A and B). Nonenhancement was observed on the initial MR studies. Diffuse gyral enhancement was observed on contrast-enhanced T1-weighted images obtained some weeks after the onset of symptoms, which revealed blood-brain barrier (BBB) disturbance (cases 1 and 2). Furthermore, an increase in the permeability of the BBB can facilitate the passage of CsA and FK 506, thus promoting cerebral ischemia and extending neurologic damage. The development of cortical laminar necrosis with cortical hyperintensity and diffuse cortical atrophy on T1-weighted images may indicate that a hypoxic episode has occurred (Figs 1E, 2D, and 3E). On the other hand, some of the lesions observed in these two patients had an anastomotic border zone distribution (Figs 1B and 2A). Therefore, we agree with Bartynski et al (13) that the watershed distribution of the lesions, without anastomotic border infarction, supports the theory of hypoperfusion or ischemia produced by major and minor vessel injury.
Our third patient had clinical symptoms and neuroradiologic findings similar to the other two patients. She, however, was not treated with immunosuppressive drugs, but rather chemotherapy (vincristine and methotrexate). To our knowledge, this is the first reported case of cortical laminar necrosis related to chemotherapy treatment.
Neurotoxicity related to vincristine has been well documented (4, 5). The most common side effects are peripheral neuropathy and autonomic neuropathy. Vincristine does not pass the BBB, and therefore central neurotoxicity is uncommon and may appear with cortical blindness or the syndrome of inappropriate antidiuretic hormone secretion (4, 5, 14). In the few cases of cortical blindness related to vincristine reported in the literature, CT findings were normal and no MR imaging data were available except in the two cases described by Heran et al (14), in which bilateral symmetrical lesions in the temporo-occipital regions were present on CT and MR studies, similar to findings in CsA-related neurotoxicity. On the basis of their neuroradiologic findings, Heran et al suggested a vascular ischemic process. In our case, the cortical laminar necrosis observed on a follow-up MR imaging study also suggested a hypoxic-ischemic episode.
The pathogenesis of vincristine neuropathy is poorly defined but is probably related to an effect on microtubules. Microtubules are a major cytoskeletal element and are essential in the segregation of chromosomes during cell division, in axonal transport, in secretory process, and in the maintenance of specific cell morphology (5). The mechanism of a vascular injury could be due to direct endothelial toxicity, because vincristine has a slow plasma clearance rate and accumulates to the greatest extent in vascular tissue. Endothelial damage could provoke a BBB disturbance and perhaps a hypoxic episode.
Neurotoxicity caused by methotrexate is also well documented. Chronic leukoencephalopathies and acute transient cerebral dysfunction are due to high doses of intravenous methotrexate. The rate of acute transient cerebral dysfunction is 15% (15). Neuroradiologic studies are usually normal, although periventricular hypodensity on CT scans has been described (16).
The possible mechanisms by which methotrexate may cause neurologic disturbances are unknown. Patients do not have recurrent symptoms after subsequent chemotherapy, which suggests that this is not a hypersensitivity phenomenon (5). A biochemical factor must be considered. Methotrexate may interfere with the synthesis of nucleotides, thereby inhibiting nucleic acid biosynthesis and replicate functions. This can also produce cellular death and increase the oxidized folates, which may be toxic to the brain. Methotrexate also affects the replication of microglia and endothelial cells. Damage to these structures could result in insufficient removal of metabolic waste products and cellular debris, and also permit other neurotoxic drugs, like vincristine, to cross the BBB (15).
It is difficult to know which drug was responsible for the neurotoxicity in case 3; however, no methotrexate-related cortical blindness has been reported, and perhaps the combination of these two drugs can increase their neurotoxic effects.
Footnotes
↵1 Address reprint requests to Dra. Nuria Bargalló, Department of Radiology, Hospital Clínic de Barcelona, Villarroel, 170, 08036 Barcelona, Spain.
References
- Received June 1, 1999.
- Copyright © American Society of Neuroradiology




{kind=link}
{kind=link}
{kind=link}