
Abstract
Summary: We report a rare case of intradural primary osteosarcoma (IPOS) in a 74-year-old man with aphasia and right-sided hemiparesis. Radiologic workup revealed a large, partially calcified, left-sided frontotemporal intracranial mass lesion. At surgery, the tumor was found to be entirely intradural; it involved the brain and subarachnoid space of the left sylvian fissure. The adjacent dura was uninvolved. Neuropathologic findings confirmed the diagnosis of chondroblastic osteosarcoma. To our knowledge, this is the sixth reported case of IPOS and the first reported case of the chondroblastic subtype.
Osteosarcoma (OS) is a highly malignant neoplasm that typically arises in the appendicular skeleton of adolescents and young adults (1). Extraskeletal OS (ESOS) is a rare subgroup of OS that occurs in the soft tissues of the extremities, retroperitoneum, and trunk during the fifth and sixth decades of life. Primary intracranial OS is a rare tumor that can arise from the osseous skull base, but it also may originate from mesenchymal components such as dura. When an OS occurs in the brain or subarachnoid space, has no dural attachment, and is not metastatic, it can be considered an intradural ESOS or intradural primary OS (IPOS). These tumors are exceedingly rare. Histologic findings in the five previously reported cases (2–6) included three pleomorphic, one osteoblastic, and one small cell subtypes.
Case Report
A 74-year-old white man, with no remarkable medical history, was found in the back of his van. He was conscious but unable to speak. Neurologic examination revealed increased tone in the left extremities, right hemiparesis, and aphasia. The patient was dehydrated and had decubitus ulcers on his right shoulder, anterior chest wall, and thigh; these findings suggested he had been incapacitated for a prolonged period.
Nonenhanced CT revealed a large intracranial mass lesion with both intra- and extraaxial components in the left frontal and temporal lobe regions, with subfalcial herniation and substantial compression of the ventricular system (Fig 1A). The mass was heterogeneous in appearance, with areas of both soft-tissue and cystic attenuation. Confluent calcification was seen in the lateral aspect of the lesion, close to the left temporal bone (Fig. 1B). On MR images, the mass was multicystic in appearance with low signal intensity on T1-weighted images and high signal intensity on T2-weighted images (Fig 2A and B). Curvilinear hypointensity on T2-weighted images that corresponded with the calcification on CT scans was identified (Fig 2C). Numerous septae had intense enhancement (Fig 2D). The mass involved the left sylvian fissure and appeared to engulf the left middle cerebral artery (MCA) and its branches. The lateral margin of the tumor abutted the left temporal squamosa, and a small area of focal dural enhancement was present in the same location. Osseous or dural involvement could not be excluded on the basis of imaging findings.

Axial CT scans.
A, Nonenhanced image at the level of the foramen of Monro shows a large, left frontotemporal mass lesion with heterogeneous attenuation, including soft-tissue, cystic, and calcified components. Note the subfalcial herniation and mass effect.
B, Scan shown in A, but obtained with bone window settings, depicts curvilinear calcifications in the lateral aspect of the mass.
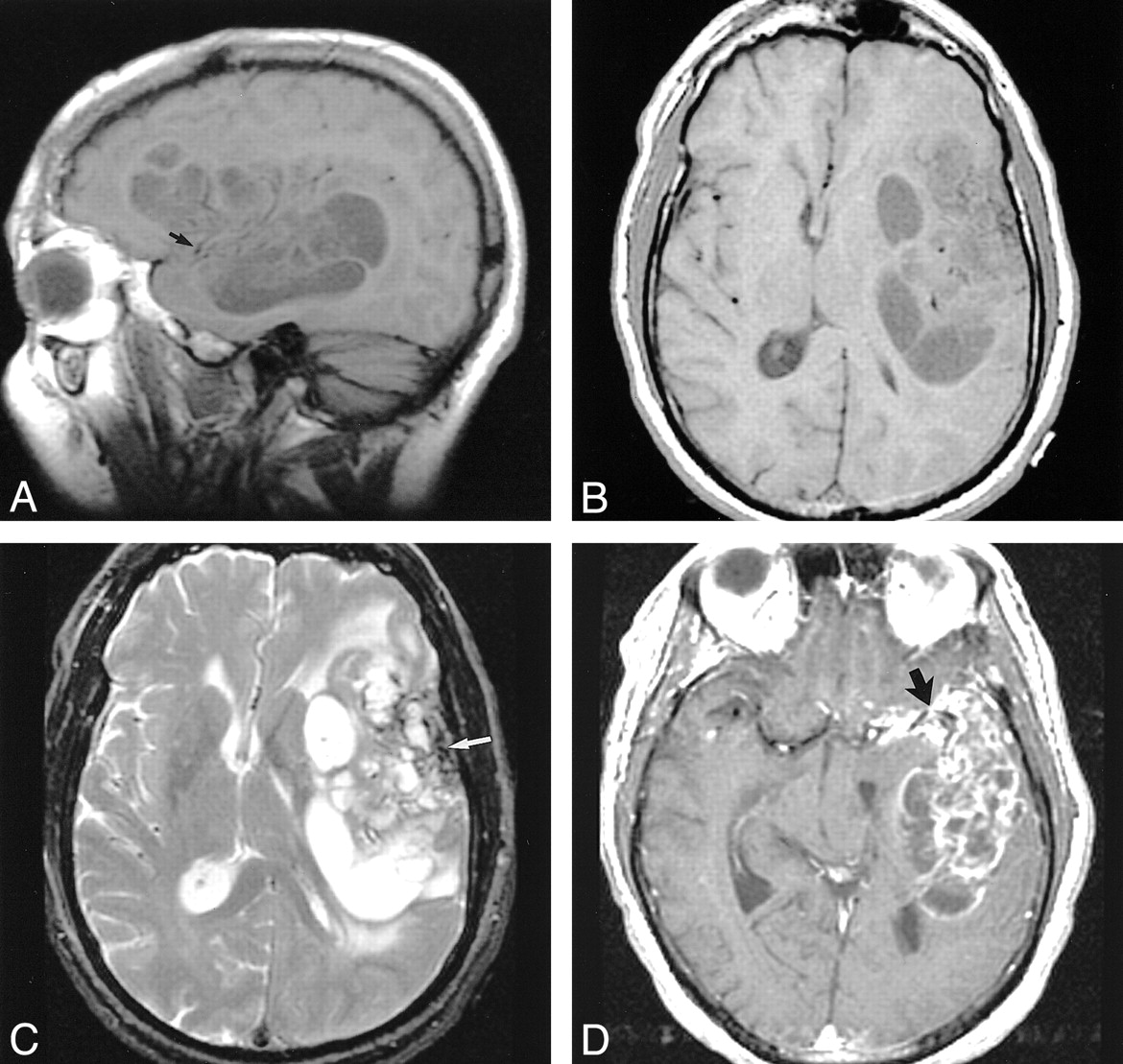
MR images.
A and B, Sagittal (A) and axial (B) T1-weighted images (600/17 [TR/TE]) show the extent of the left frontotemporal mass. Focal areas of hypointensity are separated by isointense septa. Punctate flow voids of the left MCA are surrounded by abnormal signal intensity (arrow in A).
C, Axial T2-weighted image (2600/90) reveals focal areas of hyperintensity corresponding to the hypointensity in A and B. Septa remain relatively isointense. Curvilinear hypointensity in the lateral aspect of the mass represents calcification (arrow).
D, Axial contrast-enhanced T1-weighted image (600/17) demonstrates strong enhancement in the septa within the mass. Also note the more confluent enhancement encasing the left MCA bifurcation in the sylvian fissure (arrow).
At surgery, frontotemporoparietal craniotomy was performed on the left side. The dura, which was opened without difficulty, was not adherent to the underlying brain or tumor. Calcified tumor was seen on the surface of the brain. A circumferential dissection was performed to debulk the lesion. The mass was found to have focal areas of hemorrhage, calcification, cartilage, and cystic-necrotic components. A considerable amount of tumor was in the sylvian fissure; it was not removed because of its intimate involvement with the MCA. A radiographic bone survey and nuclear medicine bone scanning were performed postoperatively to assess for an occult primary. None was found. The patient's postoperative course was tumultuous, and the patient died 10 d after admission.
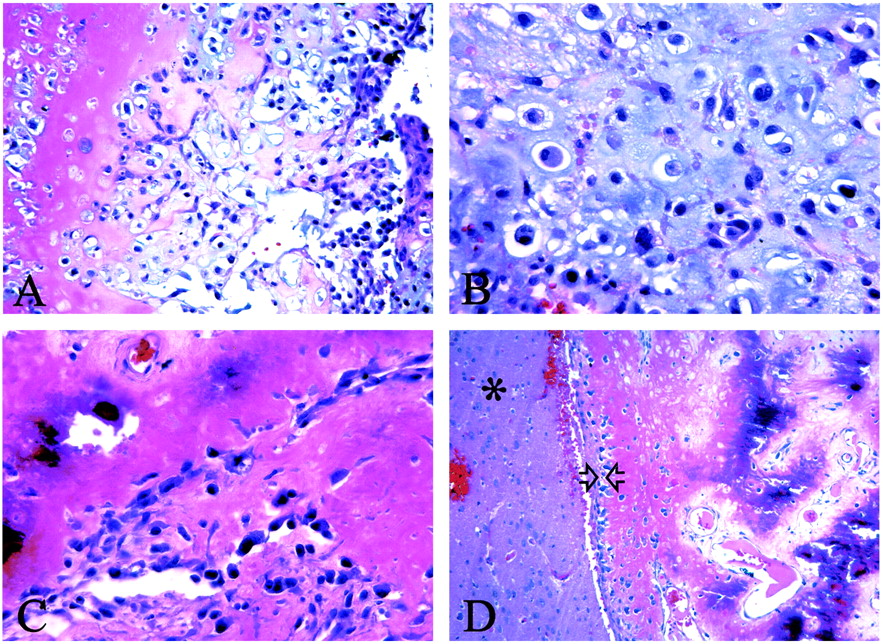
Pathologic gross description revealed gray matter–white matter soft tissue with cystic and calcified areas. Microscopic sections revealed a moderately cellular neoplasm that had moderate to marked cellular atypia. Occasional mitotic figures were seen. Areas of osteoid matrix and immature bone were identified. In addition, large areas of neoplastic cartilage were present. The tumor had a broad expansile interface with the surrounding gliotic brain. No evidence of a glial tumoral component was present (Fig. 3A–D). The final neuropathologic diagnosis was high-grade chondroblastic OS.

Photomicrographs show that the neoplasm involves both bone and cartilage.
A, Neoplastic chondrocytes in a chondroid matrix (pink) are adjacent to osteoblasts (purple) (hematoxylin-eosin, magnification × 200).
B, Higher-power view of an area of bone differentiation shows that atypical neoplastic osteoblasts are associated with a partially mineralized osteoid stroma (hematoxylin-eosin, magnification × 400).
C, Higher-power view of the chondrosarcoma component shows that atypical chondrocytes are scattered within a somewhat immature chondroid matrix (hematoxylin-eosin, magnification × 400).
D, The tumor has an expansive interface (arrowheads) with the surrounding gliotic brain parenchyma (asterisk) (hematoxylin-eosin, magnification × 400).
Discussion
ESOS is an uncommon malignant neoplasm that accounts for approximately 4% of all osteosarcomas and 1.2% of all soft-tissue sarcomas (7, 8). ESOS typically occurs in patients older than 30 y, with a peak incidence in the sixth decade of life. The most common locations include the lower extremities, upper extremities, and retroperitoneum. The histologic features of ESOS are the same as those of traditional OS; ESOS has osteoblastic, fibroblastic, and chondroblastic subtypes. Less common subtypes include small cell, giant cell, and telangiectatic subtypes. The prognosis is uniformly poor, and no standardized treatment protocol exists.
Primary OS (POS) of the CNS is rare; it can be divided into those arising from bone or dura and those that are entirely intradural. In a recent review of the literature, Ashkan et al (9) described 19 de novo cases of POS, including 14 arising from bone, four originating from dura, and one that could not be classified further because it involved both skull and dura. Also noted were seven cases of secondary intracranial OS arising from bone in the setting of prior radiation therapy.
IPOS is exceedingly rare, and to our knowledge, only five cases (2–6) have been cited in the modern literature. Cannon et al (10) recently reported the only case of IPOS arising in the cerebellum; however, that lesion was associated with, and possibly arose from, an epidermoid cyst. In 1976, Jacques et al (2) reported the first case of IPOS in a 53-year-old man. The microscopic description suggests that this was a pleomorphic OS. The IPOSs reported by Reznik and Lenelle in 1991 (3) and Bauman et al in 1997 (6) also had a pleomorphic histologic finding with no dominant cell type. The osteoblastic and small cell subtypes have each been reported once (4, 5). To our knowledge, this is the first reported case of the chondroblastic subtype of IPOS.
On the basis of preoperative imaging findings, a differential diagnosis that included mesenchymal sarcomas of the adjacent dura or calvaria, including chondrosarcoma and OS, was offered. No history of previous radiation therapy or evidence of coexistent Paget disease suggested a secondary sarcoma. Although a direct attachment to the skull base was not identified at imaging, the multicystic appearance of this mass and septal enhancement somewhat suggested a chondrosarcoma at the skull base. Statistically, an IPOS was not seriously considered. A meningioma variant or hemangiopericytoma was not seriously considered, because relatively little dural thickening or enhancement relative to the size of the intracranial component was present. A metastatic lesion was excluded on the basis of thorough radiologic assessment. The signal intensity characteristics and enhancement pattern were not consistent with those of an epidermoid cyst. At surgery, a clean plane between the dura and tumor was identified. The dura was not sent for pathologic evaluation, because it was observed to be uninvolved at surgery. The tumor was described as having hard calcification, as having cartilaginous areas, and being a “suckable tumor.” This description can be applied to the many skull base chondrosarcomas that are surgically excised at this institution, and it correlates with the neuropathologic description of a chondroblastic dominant subtype.
The origin of this tumor is not clear. It has been postulated that the sarcomatous component of gliosarcomas arises from mesenchymal elements associated with perivascular sheaths (such as fibroblasts, endothelial cells, smooth muscle cells, and/ or pericytes) or from the arachnoid (11–13). In the case that Reznik and Lenelle (3) described, a thalamic mass extended into the peripeduncular arachnoid and surrounded a medium-sized artery. Bauman et al (6) described tumoral involvement in the right MCA. In the current case, a large portion of tumor was intimately involved with the left MCA in the sylvian fissure, which precluded further debulking. Given the presumed origin of the sarcomatous elements of a gliosarcoma, we postulate that the current tumor arose from mesenchymal components in the sylvian fissure or along the perivascular sheath of a blood vessel. The exact cause of these rare lesions remains uncertain.
Footnotes
1 Address reprint requests to Matthew T. Walker, MD, Northwestern Memorial Hospital, Northwestern University Medical School, Department of Radiology, Section of Neuroradiology, 676 N. St. Clair St., Chicago, IL 60611.
References
- Received April 18, 2001.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.