
Abstract
Summary: We present the case of an 11-year-old female patient with tuberous sclerosis who had a right nasal mass. CT examination revealed fibrous dysplasia involving the frontal, ethmoid, sphenoid, and vomer bones. Biopsy findings of the mass confirmed this diagnosis, and follow-up revealed marked expansion of these lesions. The authors emphasize the association of bone abnormalities and tuberous sclerosis and discuss the consideration of fibrous dysplasia as a component of this syndrome.
Tuberous sclerosis (TS) is a disease of autosomal dominant inheritance classically characterized by a clinical triad of epileptic seizures, mental retardation, and skin lesions (1). Three CT findings of skull changes associated with TS are: patchy areas of increased bone attenuation, generalized thickening and increased attenuation of both tables of the skull vault, and evidence of elevated intracranial pressure (eg, suture diastasis, sellar changes, increased convolutional markings) (2).
Fibrous dysplasia is a disorder characterized by progressive replacement of normal bone elements by fibrous tissue involving the skull and facial bones in 10–25% of cases of monostotic form and 50% of patients with the poliostotic variety (3, 4). A search of the literature shows only one previous report that describes the association between TS and fibrous dysplasia involving the skull (5). Herein, a case of TS and fibrous dysplasia that involves the cranio-facial bones is presented.
Case Report
A 2-year-old female patient was evaluated for a 1-year history of epileptic seizures. At birth, a hyperpigmented nevus was on the thorax, and small pigmented papules were observed in the malar region. The patient had normal psychomotor development until 1 year of age, when developmental delay started. Segmental examination revealed angiofibroma over the malar regions, shagreen patches on the back, and hypopigmented macules on the thorax. Ophthalmologic evaluation showed amaurosis and retinal hamartomas on the right eye, and neurologic examination was remarkable only for mild mental retardation. Eletroencephalography revealed a pattern compatible with West syndrome, and a cranial CT scan showed several subependymal nodules on the lateral ventricles and cortical tubers in the brain hemispheres. Seizure medication was started (valproic acid, 1 mL). Investigation of other abnormalities associated with TS was unremarkable, except for an abdominal sonogram showing a left renal mass, which was histologically confirmed to be an angiomyolipoma.
Five years after the initial presentation, a CT scan showed thickening of the calvarium with opacification of the diploic space on the right fronto-temporal region (Fig 1). Two years later, the patient presented with a right nasal mass, which was observed on physical examination. The CT scan revealed marked osseous abnormalities on the frontal bone with ground-glass appearance and ethmoid, sphenoid, and vomer bones with a cystic pattern, which were typical of fibrous dysplasia. These lesions were predominantly on the right side (Figs 2 and 3). A biopsy of the nasal mass was performed, and histologic examination confirmed what imaging findings had suggested. On follow-up, extension of the cranio-facial bone lesions was observed, and surgical resection and reconstruction was indicated.
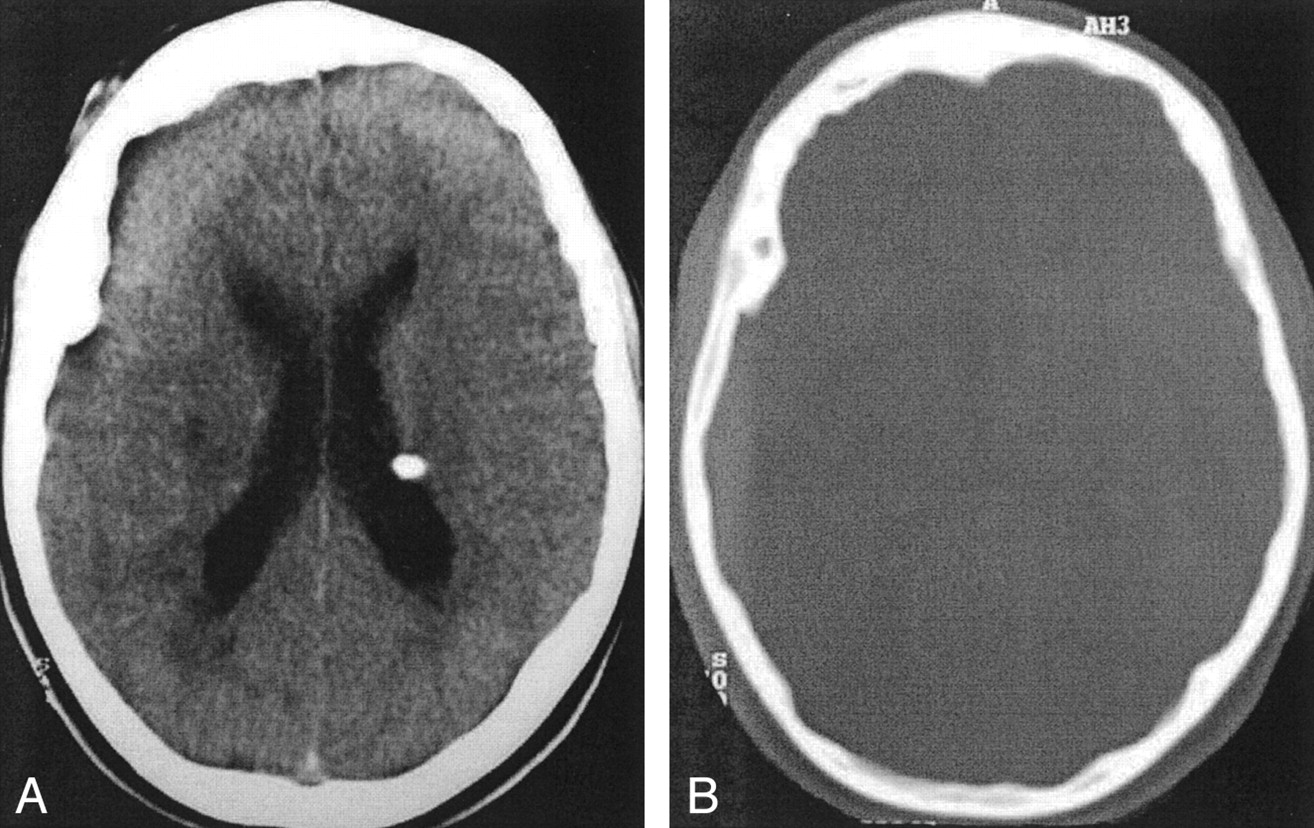
Axial cranial CT scan (A) shows subependymal nodules in the lateral ventricles and a tuber at the right internal capsule. Another axial CT scan (B) shows thickening of the bone at the right fronto-temporal region.
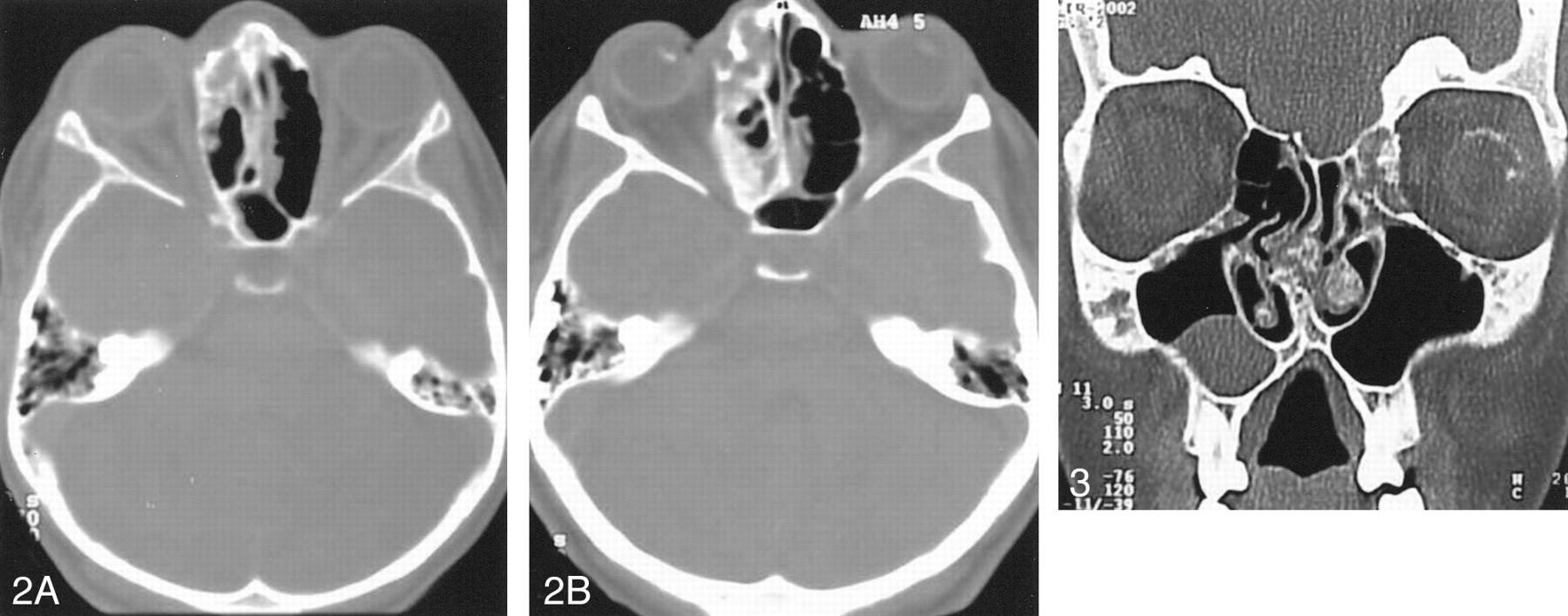
Axial cranial CT scan (A) shows the ethmoid bone lesion with a mixture of ground-glass and cystic patterns. Four years later, a CT scan (B) obtained at the same level as that in A, shows progression of the lesion.

Coronal CT scan reveals frontal bone lesions with a ground-glass pattern and a cystic form in the vomer.
Discussion
TS is a neurocutaneous syndrome that shows a tendency to dominant inheritance and has a prevalence ranging from 1:10,000 to 1:200,000 live births. This entity was first described in 1880 by Bourneville as involving the central nervous system. The systemic involvement was demonstrated by Vogt, who described the classic triad of epileptic seizures, mental retardation, and skin hamartomas. These hamartomas may occur in many organs, with a variety of clinical manifestations. Recognition is of prime importance, because components of the triad may not appear simultaneously or may not appear at all. Furthermore, seizures, although observed in the first decade of life, are nonspecific, mental retardation is difficult to evaluate at birth, and skin lesions may be subtle or absent in early life (1, 2). In this case, the diagnosis of TS was confirmed by the age of 2 years, and the patient had facial angiofibroma, retinal hamartomas, renal angiomyolipoma, and brain lesions (cortical tubers and subependymal hamartomas) as diagnostic criteria.
Skeletal radiographic abnormalities described in TS include hyperostosis of the calvaria, osteoblastic or osteosclerotic changes in the spine, and cystic changes in the phalanges. Some CT features of skull abnormalities in patients with TS have been reported: 1) Patchy areas of increased bone attenuation in the calvaria occur after puberty and are more common in the parietal region. These changes have been shown to be due to hyperostosis of the inner table and of the trabeculae of the diploic spaces. 2) A more generalized thickening and increased attenuation may occur in both tables of the skull vault. 3) Evidence of elevated intracranial pressure (suture diastesis, sellar changes, increased convolutional markings) is a rare manifestation that occurs when a subependymal hamartoma causes obstructive hydrocephalus (1, 2, 6). At 8 years of age, our patient had CT findings of thickening of the calvarium in the right fronto-temporal region.
In 1988, Breningstall et al (5) reported a case of TS and radiographic abnormalities characterized by marked sclerosis and thickening of the left wing of the sphenoid and osteosclerotic areas in the left acetabulum. Biopsy specimens of these lesions, which were characteristic of fibrous dysplasia, were analyzed, and the diagnosis of the poliostotic unilateral form of TS was confirmed. This skeletal abnormality, typically seen in adolescents and young adults, is a developmental anomaly in which normal bone marrow is replaced by fibro-osseous tissue. Histologic examination reveals a background of whorled bundles of spindle cells and multiple trabeculae that vary both in size and shape. These trabeculae are composed of immature woven bones that presumably result from osseous metaplasia. Scattered osteoclasts are also seen, but they are not a dominant histologic feature.
Fibrous dysplasia may be associated with either solitary or multiple lesions in one or more bones. Approximately 70–80% of cases are monostotic, and 20–30% are poliostotic. In most cases, few bones are involved, but virtually any bone may be affected. Monostotic fibrous dysplasia is most frequently encountered in a rib, femur, tibia, calvaria, and humerous. The poliostotic variety is observed more commonly in the skull and facial bones, pelvis, spine, and shoulder girdle. Involvement of the skull and facial bones occurs with nearly equal frequency and is noted in 10–25% of patients with monostotic form and 50% of the cases of poliostotic variety. Chong et al (3) reviewed the radiographic features of fibrous dysplasia of the skull and facial bones and described three patterns. The pagetoid or ground-glass pattern is the most commonly observed finding (56%) and consists of a mixture of dense and radiolucent areas of fibrosis. Sclerotic lesions (23%) are homogeneously dense, whereas the cystic variety (21%) is characterized by a spherical or ovoid lucency surrounded by a dense, bony shell. The expansion of these lesions over time is also a typical finding (3, 4, 7). In our patient, frontal, ethmoid, sphenoid, and vomer bones were affected and showed a mixture of ground-glass and cystic patterns. Although four bones had lesions, we intended to classify this patient’s lesion as monostotic, because it affected neighboring bones, and no other osseous lesion was observed.
The differential diagnosis of fibrous dysplasia of the skull and facial bones includes every lytic, sclerotic, or mixed lesion. The radiologic appearance may be confused with that of other conditions such as benign bone tumors, meningioma, Paget disease, and Langerhans cells histiocytosis, but CT or MR imaging of this lesion is almost always sufficient to correctly diagnose the disease (4, 7, 8).
Conclusion
Both TS and fibrous dysplasia are congenital diseases, but TS is autosomal dominant in inheritance, whereas fibrous dysplasia is sporadic in occurrence. Our case and a previous report of fibrous dysplasia of the skull associated with TS expand the association of bone abnormalities and TS and emphasize the possibility that fibrous dysplasia may be a component of this syndrome.
- Received July 24, 2002.
- Accepted after revision September 5, 2002.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}