
Abstract
BACKGROUND AND PURPOSE: Malformations of the brain stem are uncommon. We present MR imaging and diffusion tensor imaging (DTI) features of 6 patients with pontine tegmental cap dysplasia, characterized by ventral pontine hypoplasia and a dorsal “bump,” and speculate on potential mechanisms by which it forms.
MATERIALS AND METHODS: Birth and developmental records of 6 patients were reviewed. We reviewed MR imaging studies of all patients and DTIs of patient 3. Potential developmental causes were evaluated.
RESULTS: All patients were born uneventfully after normal pregnancies except patient 6 (in utero growth retardation). They presented with multiple cranial neuropathies and evidence of cerebellar dysfunction. Variable hypotonia and motor dysfunction were present. Imaging revealed ventral pontine hypoplasia and mild cerebellar vermian hypoplasia, in addition to an unusual rounded to beaklike “bump” on the dorsal surface of the pons, extending into the fourth ventricle. Color fractional anisotropy maps showed the bump to consist of a bundle of axons directed horizontally (left-right). The bump appeared, on morphologic images, to be continuous with the middle cerebellar peduncles (MCPs), which were slightly diminished in size compared with those in healthy infants. Analysis of the DTI was, however, inconclusive regarding the connections of these axons. The decussation of the MCPs, transverse pontine fibers, and longitudinal brain stem axonal pathways was also abnormal.
CONCLUSIONS: Our data suggest that the dorsal transverse axonal band in these disorders results from abnormal axonal pathfinding, abnormal neuronal migration, or a combination of the 2 processes.
Malformations of the brain stem are very rare and usually result in severe clinical manifestations: notably feeding disorders, impaired swallowing, sensorineural hearing loss, long tract signs, and palsy of cranial nerve V. They have recently been classified into several groups of disorders, including malformations with abnormal brain stem segmentation or segmental hypoplasia, postsegmentation malformations (putative pathfinding or migration abnormalities), and malformations associated with abnormal cortical organization.1 We report MR imaging and diffusion tensor imaging (DTI) findings of an unusual malformation characterized by pontine hypoplasia with ectopic dorsal transverse pontine fibers in 6 patients presenting with the clinical manifestations of multiple cranial nerve palsies and variable long tract signs. Similar MR imaging features have been described as pontocerebellar hypoplasia,2 Möbius syndrome,3 and, recently, a new malformation called “pontine tegmental cap dysplasia.”4 We discuss the clinical presentation, morphologic abnormalities, and the potential underlying mechanisms that may cause this interesting malformation.
Materials and Methods
Six patients were found in different centers, where they were evaluated in infancy for delayed development or failure to thrive. All were referred for routine brain MR imaging after pediatric neurologic examination disclosed multiple cranial neuropathies. No family history of brain malformation was obtained in any of the patients. Clinical data available for each patient are summarized in Table 1.
Patient data
All patients were examined by MR imaging at 1.5T, including axial, coronal, and sagittal T1-weighted (from direct acquisitions with 4-mm section thickness or sagittal 3D spoiled gradient-recalled-echo and magnetization-prepared rapid acquisition of gradient echo sequences [SPGR, MPRAGE] with coronal and axial reconstructions) and/or T2-weighted images (4-mm section thickness). Patient 3 and a neurologically healthy control, matched for age, underwent DTI performed on a Trio 3T MR imaging scanner (Siemens, Erlangen, Germany) sampling the data in 30 directions. Other parameters were the following: TE = 96 ms; TR = 6600 ms; b = 1000 s/mm2; FOV = 240 × 240 mm; matrix = 128 × 128; section thickness = 2 mm; number of averages = 2; bandwidth = 1396 Hz/pixel with generation of cross-sectional directionally encoded fractional anisotropy (FA) color maps.
Results
All imaging findings are summarized in Table 2. Anatomic images in all patients revealed a small pons with a variably shaped band of white matter coursing like an exophytic mass along its dorsal superior aspect (Fig 1). In patient 4, this band had a beaklike appearance, but in the other patients, it was more roundly shaped, smaller and smoother (caplike). These fibers appeared continuous with the middle cerebellar peduncles on coronal MR images (Fig 2A). In some patients, particularly patient 4 (Fig 1D), the pons appeared extremely thin immediately caudal to the band. Additional brain stem anomalies included a short mesencephalon-pontine junction (isthmus) that was thin in patient 1, and short and thickened in the others (Fig 1). In most cases, the 4th ventricle was partially occupied by the dorsal band (Fig 1B–E). The course of the superior cerebellar peduncles (SCPs) was somewhat lateral in position in patients 1 (Fig 2B) and 4. The middle cerebellar peduncles (MCPs) were quite small (Fig 2D), and the ventral pons was flattened. The medulla was small, and in patients 1 and 4, it appeared elongated (Fig 1). The inferior cerebellar peduncles (ICPs) were quite small in patient 4 and could not be identified in the others. The cerebellar vermis was normal in patient 3 and small in patients 1 and 6, whereas it was small with abnormal orientation of fissures in patients 2, 4, and 5 (Fig 1C, -D). The cerebellar hemispheres were small in patients 1, 2, 4, and 6. Axial sections showed hypoplasia of the inferior olives (not shown).
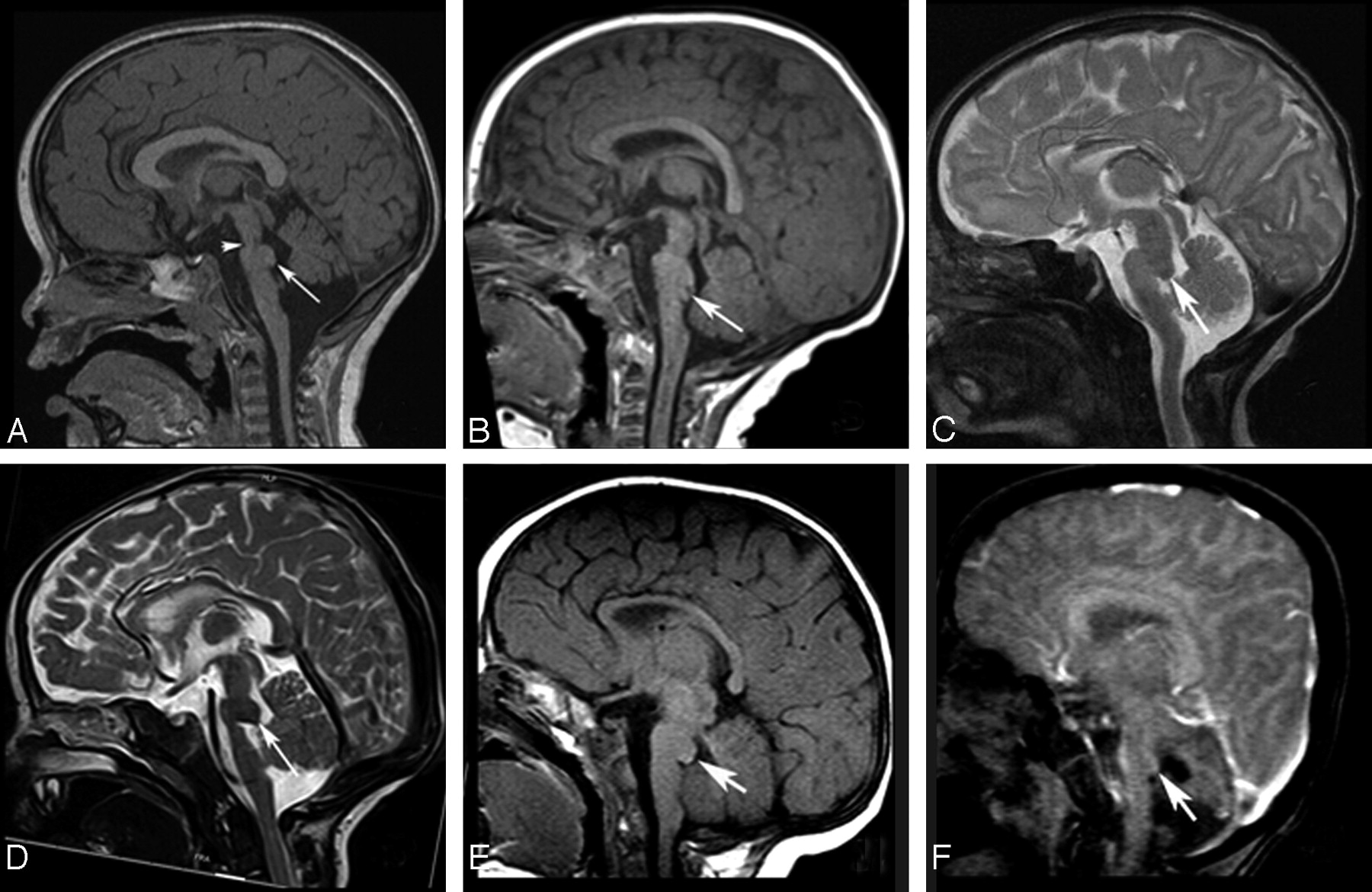
A and B, Midsagittal T1-weighted images (T1WIs) in patients 1 and 3 show flattening of the ventral pons, thinning of the isthmus (arrowhead, A), dysmorphism of the dorsal upper pons (caplike) bulging (long arrows) and protruding in the fourth ventricle (B). The vermis is hypoplastic in A. C and D, Midsagittal T2WIs in patients 2 and 4 show the same findings as in A and B, with slightly different patterns (beaklike shape in D) (long arrow). In both cases, the vermis is hypoplastic and dysplastic and the CC is hypoplastic. The CC is dysmorphic in patient 3 (B) and thin in patient 4 (D). E and F, Midsagittal T1- and T2WIs for patients 5 and 6 show the tegmental cap with hypoplastic (E and F) and dysplastic (E) vermis. F, Fourth ventricle is slightly enlarged. The CC is hypoplastic in E and dysmorphic in F.

Patient 1: A and B, Coronal T1-weighted images and axial T2-weighted images (T2WIs) show the “molar tooth” appearance of the elongated SCPs running laterally (arrows) and the dorsal band crossing the midline and likely joining the MCPs (arrowheads). C, Axial T2WI at the level of the MCPs, which appear small (white arrows). D, Axial T2WI at the level of middle pons. A “horizontal cleft” is visible, outlined by black arrowheads. The black star indicates the fourth ventricle. Note the small size of cerebellar hemispheres (CH) and the hypoplastic vermis (V).
MR imaging findings in all patients and DTI findings in patient 3
Relatively minor supratentorial anomalies were observed. The corpus callosum (CC) was dysmorphic in patients 1 and 6 but was thin in patient 3 and hypoplastic in the others. Cerebral hemispheres, olfactory bulbs, orbits, optic pathways, hypothalamic areas, and pituitary glands were structurally normal. There was no evidence of a supratentorial cortical malformation in any of the patients. Pineal cysts were found in patients 1 and 3, the latter showing, in addition, a Rathke cleft cyst; these were considered to be incidental findings. Segmentation anomalies of the spinal column, with associated scoliosis, were found in 2 patients. Orbits, calvaria, skull base, and visualized inner and middle ear structures appeared normal in all patients.
FA maps and fiber tractography were obtained in patient 3 (Fig 3) and were compared with studies obtained in the control patient. Cross-sectional FA images verified the impression from the anatomic images that the MCPs were small. The decussation of the MCPs and the fibers to the pontine nuclei were absent at the level of the upper pons, likely explaining the decreased size of the ventral pons. At this same level, an abnormal transverse bundle of axons crossed the midline in the dorsalmost pons, extending from 1 MCP to the other to form the pontine “cap.” On the FA and tractography images, these fibers were not obviously continuous with any white matter bundle; indeed, they appeared to terminate at the sites of crossing of the craniocaudally oriented SCPs (Fig 3B, -C). The SCPs appeared elongated and running laterally. Their decussation was not visible in its normal midbrain location. The ICPs were not visible. Two tracts of longitudinal axons were located in the anterior pons, at the sites normally occupied by the corticospinal and corticopontine tracts, though the size of these bundles of axons was smaller than those seen in the healthy control. It could not be determined whether the dorsal longitudinal axonal pathways (the median longitudinal fasciculus, the medial and lateral lemnisci, etc) were absent or included in these more ventral bundles.

Patient 3: A and B, Color FA cross-sectional images show elongation of the SCP (green).The white star indicates the fourth ventricle. Descending long tracts (corticospinal and corticopontine) appear in blue. The ventral and middle transverse pontine fibers are missing; the SCP decussation is not visible. B, The ectopic bundle of fibers appears in red at the dorsal aspect of the pons and seems not to connect the MCPs visible laterally. C, Coronal FA image shows the elongated SCPs running laterally (arrows); the dorsal band (arrowheads) crossing the midline may join either the SCPs vertically or the MCPs horizontally. D and E, 3D projections of tractography. In the control (D), ventral transverse fibers are clearly seen (black arrow) and the MCPs are of normal size (arrowheads). In patient 3 (E), long descending tracts and the MCPs appear smaller (arrowheads). The ectopic dorsal pontine fibers are visible (long arrow) and are not seen to definitely connect the MCP (arrowheads).
Discussion
Malformations of the brain stem are not well described in the medical literature. One reason is that the brain stem is poorly evaluated by both pathology and neuroimaging. Pathologic analysis of the malformations is inhibited by difficulty in evaluating the axonal pathways using traditional techniques of brain cutting. Early MR imaging was hampered by long imaging times, artifacts generated by rapid CSF flow in the perimedullary and peripontine cisterns, and the inability to differentiate nuclei from white matter pathways. Only recently, with the development of parallel imaging techniques (to improve signal intensity–to-noise ratio and the speed of imaging), stronger gradients (to allow thinner imaging sections), and DTI (to allow evaluation of white matter tracts) has MR imaging begun to be useful in the evaluation of brain stem disorders.5–7 We report here the MR imaging evaluation of 6 patients and DTI of 1 with an unusual malformation of the pons, which may be more common than generally realized.
Similar MR imaging findings have been previously reported2,3 but were not recognized as a unique entity until Barth et al4 described 4 cases, which they called “pontine tegmental cap dysplasia.” It has been reported in sporadic male and female cases without affected siblings or consanguinity. Although autosomal recessive inheritance cannot be excluded, new dominant mutations, such as submicroscopic chromosomal rearrangements, may be a more likely mechanism.8 To date, to our knowledge, none of the published patients have been evaluated with tiling-path or high-attenuation oligonucleotide comparative genomic hybridization arrays.
Our morphologic and DTI analyses of the affected brain stem gave results that are similar to those reported by Barth et al.4 The band on the dorsal aspect of the upper pons appears to be a bundle of transversely oriented fibers (red on the color FA map, Fig 3B). Although the axons appear to be continuous with the MCPs on the morphologic image (Fig 2A, -D), this impression is not confirmed by the color FA map, where the fibers seem to terminate when they reach the SCPs (Fig 3C, -D). However, connectivity may not be demonstrated because of the technical limitations of DTI (tractography terminates when either the FA or the angle of the tract falls below a specified threshold). Therefore, just as the corticospinal tracts often appear to terminate in the cerebrum immediately above the lateral ventricles, where the perpendicular callosal fibers cross and reduce intravoxel anisotropy to nearly zero, the pontine cap axons, coursing horizontally, may appear to terminate when crossing the craniocaudally oriented SCPs.
Tractography, by using higher angular resolution diffusion techniques that can better follow axons through regions of complex axonal anatomy (Q-ball imaging, spheric deconvolution, or persistent angular structure) might better resolve these tracts and determine the full course of the pathways in which the band is involved.5–7 Another possibility for the origin of the axons is a misorientation of the descending pathway of dorsal long tracts: medial lemniscus, lateral lemniscus, medial longitudinal fasciculus, posterior longitudinal fasciculus, central tegmental, trigeminothalamic, spinothalamic, and tectospinal tracts.1,9 Neurologic testing of these tracts was not possible in our patient because of his young age, but some of our other patients, and those described by Barth et al,4 did show some abnormalities of horizontal gaze.
The long tracts visible in the ventral pons at the level of the dorsal “cap” corresponded reasonably well, in terms of location and their rostral pathway, to the corticospinal and corticopontine tracts but were smaller than those in the control. The smaller size of the tracts might explain, at least partly, the motor problems observed in our patients. Three other major findings were identified on the color FA map of our patient: absence of the SCP decussation, markedly diminished size of the MCPs, and poor or nonvisualization of the ICPs. These observations suggest an impairment of axonal navigation, with absence or diminished number of axons, abnormal course (failure to cross the midline), and ectopic white matter fibers within the pons, as 1 possible cause of this malformation. Axonal guidance molecules such as netrins, ephrins, semaphorins, and their receptors may, therefore, be candidates for causative molecules.10–18
Migrating axons navigate by responding to chemical guidance cues via multiple membrane-associated signaling molecules. Studies of animal models have helped in identifying the molecules involved in this process. Netrin-1 interacts with DCC (attraction and sometimes repulsion) and with UNC-5 (repulsion) family receptors, which are located on the surface of the growth cone. The guidance response is assured by multiple cytoskeletal proteins.14 An aberrant trajectory could thus result from an erroneous extracellular target or a defect in proteins of the cytoskeleton. Axon guidance by netrins needs to be regulated in mice by a signaling system from the midline mediated by Slit (Sli) and its receptors Robo1, 2, and 3.19,20 An additional signaling system for Netrin (Net) uses the receptor Frazzled (Fra), which functions as a midline attractant for the tracts at the point of midline crossing.20 A loss of Net-Fra or overexpression of Sli-Robo function that results in long tracts shifting far from the midline could explain the absence of SCP decussation and the presence of a molar toothlike midbrain appearance in some of our patients, as well as the dorsal transverse axonal band.
The presence of a molar toothlike midbrain might also suggest that the primary problem in this disorder is one involving ciliary proteins, as appears to be the case in Joubert syndrome and related disorders (the “molar tooth malformations”),21–23 in which the decussation of both the SCPs and the corticospinal tracts seems to be affected.24 No renal cysts or cystic degeneration has, as yet, been reported in patients with dorsal transverse pontine bands, but the kidneys may not yet have been adequately assessed. If extra-central nervous system anomalies, such as retinal dystrophy, cystic renal disease, and polydactyly, were to be discovered, mutations affecting ciliary proteins might be revealing.
Abnormal migration of pontine neurons might also be implied by the flattened pons and by the aberrant origin of the transverse tract (presumably arising from ectopic pontine neurons) in the tegmentum. One of the Netrin-1 receptors, Unc5h3, is also involved in neuronal-migration guidance.11 Thus, a loss of Unc5h3 function may result both in abnormal neuronal migration and axonal navigation impairment. Moreover, developing neurons undergo apoptosis if synaptic connections are not established. Therefore, we can hypothesize that impaired proliferation or abnormal migration of cortical and pontine neurons might be responsible for the small size of the ventral longitudinal tracts (corticospinal and corticopontine) and one of the reasons for the flattening of the ventral pons and, in some cases as well, for the small/dysplastic vermis and small cerebellar hemispheres (patient 1). Indeed, a unifying feature in this disorder appears to be the involvement of rhombic lip (RL) derivatives: pontine nuclei, inferior olivary nuclei, cerebellar cortex (granule cells), and deep cerebellar nuclei; all arise from RL. During normal development, the transverse pontine fibers normally connect the pontine nuclei to the contralateral cerebellar cortex via the MCPs (Fig 4).25–27 The pontine cap could form by reduced ventral migration of pontine nuclei, followed by effective axon migration across the midline, leaving connections with the cerebellar cortex somewhat preserved. Another hypothesis might be that increased radial migration of pontine nuclei would result in pontine gray neurons being located near the ventricular surface of the pontine tegmentum and the consequent aberrant position of their axons. Finally, normal migration of pontine neurons followed by abnormal axon guidance away from the ventral surface of the pons is another plausible hypothesis (Fig 4D). Ultimately, gross and microscopic neuropathologic analysis of autopsy specimens or development of an animal model will be required to clarify this question. Barth et al4 performed no pathologic analysis of their patients and found no pathogenic mutations in the coding regions of the NTN1 and DCC genes in their patients.4 They also concluded that Robo3 mutations are unlikely to be causative of this complex malformation on the basis of the lack of similarity to the human disorder, horizontal gaze palsy, with progressive scoliosis known to be caused by these mutations.28

Hypotheses to explain the aberrant dorsal position of transverse pontine fibers. A, Normal development. 1) Pontine gray neurons are produced from progenitors in the rhombic lip (blue circles). 2) Newly generated neurons migrate toward the ventral midline (yellow), the “anterior extramural migratory stream” of Altman and Bayer.25 3) Axonal growth cones cross the ventral midline, leaving most neuronal cell bodies uncrossed.26 4) Cell bodies migrate toward the ventricle along radial glia.27 5) Axonal growth cones migrate into the MCP (pink). The transverse pontine fibers are sandwiched between the concurrently growing corticospinal tract (CST) and central tegmental tract (CTT). B, Hypothesis 1: Decreased ventral migration (step 2) leaves the pontine gray neurons scattered along the lateral pontine surface. Axonal growth cones cross the midline directly toward the contralateral MCP. C, Hypothesis 2: Increased radial migration (step 4) deposits pontine neurons near the ventricular surface of the pontine tegmentum. D, Hypothesis 3: Normal migration of pontine neurons. Note abnormal axon guidance away from the ventral surface, followed by midline crossing and attraction toward the MCP.
Another clue to the molecular etiology of this condition may come from the anomalies of vertebral segmentation found in 2 of our patients and 3 of the patients reported by Barth et al.4 A combination of hindbrain anomalies, cranial nerve dysfunction, and vertebral anomalies may suggest changes in function of the highly conserved Hox genes, which encode transcription factors within a deoxyribonucleic acid–binding homeodomain. The expression of genes from paralogous clusters of Hox genes in the vertebrate presomitic mesoderm results in the segmental formation of somites, from which the vertebrae derive.29 Similar Hox gene expression in the developing vertebrate hindbrain, from which the pons, medulla, and cerebellum develop, results in the segmental formation of rhombomeres, the patterning of cranial nerves, and their distribution to structures of the head and neck.30,31 Thus, abnormalities of Hox genes could be another mechanism by which these malformations develop. Again, careful pathologic analysis and development of animal models may be useful in gaining a better understanding of the causative mechanisms.
Conclusions
We report 6 new cases of pontine tegmental cap dysplasia, raising the number of all published cases of this malformation to 10.2–4 The large number of reports published recently suggests that the disorder may be more common than generally realized and may have been misdiagnosed as pontocerebellar hypoplasia or Möbius syndrome. Some of us are currently reviewing all of our pontocerebellar hypoplasia cases to determine the proportion that may actually have pontine cap dysplasia. Our findings support the hypothesis of a complex pontine malformation featuring an impairment of axonal navigation or migration of neurons generated in the rhombic lips. The etiology is unknown, but likely to be genetic. Further clinical description and neuroimaging, in combination with neuropathology and animal studies, will be required to understand this new entity.
Acknowledgments
We thank Dr. Lee Coleman for referral of patient 2.
References
- Received May 22, 2008.
- Accepted after revision August 4, 2008.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Pontine tegmental cap dysplasia: the role of diffusion tensor imaging
- Dandy-Walker Phenotype with Brainstem Involvement: 2 Distinct Subgroups with Different Prognosis
- Medullary Tegmental Cap Dysplasia: Fetal and Postnatal Presentations of a Unique Brainstem Malformation
- Tractography of the Cerebellar Peduncles in Second- and Third-Trimester Fetuses
- Brain Stem and Inner Ear Abnormalities in Children with Auditory Neuropathy Spectrum Disorder and Cochlear Nerve Deficiency
- Pontine tegmental cap dysplasia: A hindbrain malformation caused by defective neuronal migration
- Human Genetic Disorders of Axon Guidance