
Abstract
SUMMARY: Since its initial description, there have been significant changes in the epidemiology, pathogenesis, and clinical and imaging manifestations of JCV infection of brain. The most common clinical manifestation is PML. Other recently described CNS manifestations are JCE, JCVGCN, and JCM. Although AIDS is the most common predisposing factor for JCV reactivation, there is increasing incidence of brain manifestations of JCV reactivation in non-HIV settings, including different rheumatologic, hematologic, and oncologic conditions; monoclonal antibody therapy; transplant recipients; primary immunodeficiency syndromes; and even in patients without any recognizable immune deficiency. IRIS may develop secondary to restoration of immunity in HIV-positive patients with PML receiving antiretroviral therapy. This is of profound clinical significance and needs to be diagnosed promptly. Imaging plays a crucial role in the diagnosis of the disease, monitoring of treatment response, identifying disease progression, and predicting prognosis. In this article, current understanding of the epidemiology, pathogenesis, clinical presentations, and all aspects of imaging of JCV infection of the brain have been comprehensively reviewed.
Abbreviations
- ADC
- apparent diffusion coefficient
- CNS
- central nervous system
- cPML
- classic PML
- Cr
- creatine
- CTL
- cytotoxic T-lymphocytes
- DWI
- diffusion-weighted imaging
- FLAIR
- fluid-attenuated inversion recovery
- HAART
- highly active antiretroviral therapy
- HIV
- human immunodeficiency virus
- ICL
- idiopathic CD4 lymphocytopenia
- iPML
- inflammatory PML
- IRIS
- immune reconstitution inflammatory syndrome
- JCE
- JCV encephalopathy
- JCM
- JCV meningitis
- JCV
- JC virus
- JCVGCN
- JCV granular cell neuronopathy
- mAb
- monoclonal antibody
- mIns
- myo-inositol
- MS
- multiple sclerosis
- MTR
- magnetization transfer ratio
- NAA
- N-acetylaspartate
- NIRIS
- neuro-IRIS
- PCR
- polymerase chain reaction
- PML
- progressive multifocal leukoencephalopathy
- PML-IRIS
- PML associated with IRIS
- RR
- regulatory region
- SLE
- systemic lupus erythematosus
The JCV, a member of the Polyomaviridae family, was first isolated from the brain of a patient with Hodgkin disease in 1971,1 though the disease was first described by Åström et al in 1958.2 The demyelinating encephalopathy caused by the virus was subsequently termed “PML”. Until the 1980s, PML was considered an extremely rare opportunistic infection. The HIV pandemic led to a new population of immunosuppressed patients, and the prevalence of PML increased dramatically. Now, HIV-induced immunodeficiency is the most common predisposing factor for symptomatic JCV infection. The surge in HIV-associated PML has led to intense research on JCV infection and resulted in better understanding of its changing epidemiology and expanding clinicopathologic spectrum. Similarly, the imaging manifestations of JCV infection are now known to be more diverse and complex and are significantly altered by novel treatments for HIV and PML. This article presents a comprehensive review of the JCV infection, including PML, and emphasizes the broadening radiologic spectrum related to this infection.
Epidemiology: Population at Risk
It has become evident that PML has outgrown its name. JCV infection is no longer a 1-dimensional opportunistic infection, limited to HIV and lymphoproliferative disorders. Although HIV accounts for approximately 80% of the PML cases, there is increasing incidence of the disease in non-HIV settings.3
JCV is a ubiquitous human pathogen, and both inhalation and ingestion of contaminated water have been suggested as major modes of transmission of the virus.4,5 The primary infection is presumably asymptomatic, and 85% of the adult population has antibodies against JCV, implying previous exposure and potentially latent infection.6 Usually, severe deficiency of T-cell immunity (cellular immunity) is necessary for reactivation of JCV.6 Suppression of cellular immunity, secondary to HIV infection, is the major cause of the JCV reactivation and constitutes approximately 80% of patients with PML. Other less common immunodeficiency settings for PML are hematologic malignancies (13%), organ transplant recipients (5%), and autoimmune diseases treated with immunomodulators (3%).6 Epidemiology of JCV infection is described with reference to PML as this is the most common manifestation of JCV infection.
PML in HIV Infection
At present, immunodeficiency secondary to HIV-1 infection/AIDS is the most common precipitating condition that leads to JCV reactivation and PML. There are only a handful of reports of PML in the setting of HIV-2.7,8 This discrepancy may be due to variable geographic prevalence of HIV-2 infection, which is distinctly more common in African compared with Western countries.7 Sophisticated technologies required for confirmatory diagnosis of PML are not readily available in most developing countries, and the incidence may be artificially reduced accordingly.
HAART has become a central component in the therapy for HIV, leading to markedly improved survival times. HAART has substantially reduced the incidence of PML.9 In the pre-HAART era, PML affected 3%–7% of patients with HIV-1 infection and was the cause of up to 18% of fatal CNS disease.10,11 The incidence of PML has decreased from 0.7 per 100 person-years of follow-up in 1994 to 0.07 in 2001–2002.11 Unlike many other CNS opportunistic infections, JCV infection occurs early in the course of AIDS with CD4 cell counts >200/μL and can also occur in patients receiving HAART.12 One-year survival time in HIV patients with PML has also increased considerably, from 0% to 30% in the pre-HAART era to 38%–62% with HAART.13,14 However, according to a 2005 survey, PML is still the second most common cause (14%) of all AIDS-related death, second only to non-Hodgkin lymphoma.14
PML in Hematologic and Oncologic Conditions
PML was originally described in patients with chronic lymphocytic leukemia and Hodgkin disease in 1958.2 Garcia-Suarez et al15 reviewed all the reported cases of PML in lymphoproliferative disorders published between 1958 and 2004. In this extensive review, PML was linked to chronic lymphocytic lymphoma, Hodgkin disease, non-Hodgkin lymphoma, Waldeström macroglobulinemia, multiple myeloma, and mycosis fungoides. Major risk factors for PML in this setting were uncontrolled Hodgkin disease, treatment with purine analogues, and stem cell transplantation.
PML in Organ Transplantation
Organ transplantation, a setting of iatrogenic immune deficiency, is not uncommonly associated with PML. Median time to onset of the disease is 17 months, somewhat longer in patients with renal transplants due to less intense immune suppression.16 PML has also been described in patients with stem cell transplantation, both autologous and allogenic.17
PML in Rheumatologic Conditions
Calabrese et al18 recently reviewed 37 cases of PML in the setting of rheumatic diseases. All patients in this series were treated with some form of immunosuppressant before PML manifestation. Of all the rheumatologic conditions SLE was most commonly associated (65%) with PML. Other associated rheumatic diseases were rheumatoid arthritis, Wegener granulomatosis, dermatomyositis, polymyositis, and scleroderma. There are also reports of PML in patients with Sjogren syndrome19 and sarcoidosis20 with no prior immunomodulator therapy. In both of these cases, there was associated lymphocytopenia. It is unclear whether the rheumatologic condition or the lymphocytopenia was responsible for PML.
PML in the Setting of mAb Therapy
In recent years, mAbs are being used in a wide spectrum of immunologic diseases. Some of the mAbs depress the immune system and, as a result, predispose the patient to PML. Association of natalizumab (an mAb against the α4-integrin of the cell adhesion molecule family, used primarily to treat MS and Crohn disease), and PML has been extensively discussed in the medical literature.21–23 Other mAbs associated with PML are efalizumab24 (an mAb that binds to CD11a, used primarily to treat psoriasis) and rituximab25 (an mAb against CD20 used in many clinical conditions). Approximately 57 cases of PML have been described in patients receiving rituximab for the treatment of hematologic malignancy (predominantly non-Hodgkins lymphoma) (n = 50), rheumatoid arthritis (n = 1), SLE (n = 2), and autoimmune hematologic disorder (n = 4).26,27
PML in Idiopathic Immune Deficiency Syndrome
PML has also been described in patients with primary immunodeficiency disorders, ICL being the most common condition to be associated with PML.28–30 PML has also been described in patients with common variable immune deficiency.31,32
PML in the Setting of Minimal/No Immunodeficiency
Until recently, severe depletion of cellular immunity was considered an absolute requirement for the development of PML. However, there are now case reports of PML with less overt immunodeficiency, such as cirrhosis, renal failure, psoriasis, dermatomyositis, and even pregnancy.6 Furthermore, there are multiple reports in the literature of PML without any documented immunodeficiency.6 It is very important that neuroradiologists be aware of this expanding demography of PML, JCE, JCVGCN, and JCM. So far, both JCE and JCVGCN have been described in the setting of HIV infection/AIDS. Interestingly, all the reported cases of JCM have been reported in non-HIV settings including SLE and even immunocompetent patient as elaborated in the pathogenesis section.
Pathogenesis
JCV is a double-stranded circular DNA virus and a member of the Polyomaviridae family. This is a small virus with icosahedral symmetry. The capsid contains 3 viral proteins, VP1, VP2, and VP3, of which VP1 is the most abundant and can present itself to the host immune system as a viruslike particle.33,34
The pathogenesis of PML is divided into 3 phases. The first phase is a primary clinically unapparent infection. In the second phase, the virus maintains a persistent latent peripheral infection in the urinary tract, bone marrow, and probably the spleen.35 The presence of JCV in the bone marrow and shedding of the virus in urine are well documented in asymptomatic immunocompetent carriers.36,37 The CNS has also been suggested as a potential site for JCV persistence. The third or final phase is that of reactivation and dissemination of the virus with presumed hematogenous spread to the CNS.35 The route and time when the virus reaches the CNS are not known exactly. The spread is most likely hematogenous and may either be during primary infection, during peripheral persistence phase or during reactivation of the virus when cellular immunity is impaired.38
In the phase of persistent infection, the virus harbors a stable and nonpathologic RR in its DNA between the early and late protein-coding regions called archetype. With decreased level of host T-cell immunity, there is rearrangement of this archetype RR, resulting in JCV reactivation, which leads to a lytic infection of oligodendrocytes.39 Individuals with a depressed or suppressed cellular immunity rather than suppressed humoral immunity are at particular risk for PML development. CD8+ T-lymphocytes are effector cells of cellular immunity, also known as CTL. These CTLs kill the virus-infected cells if they recognize properly processed viral epitopes (a macromolecule, or part of a macromolecule derived from the virus that is recognized by the host immune system). The presence of JCV-specific CTLs in blood or CSF reduces the risk for the development of the disease and improves prognosis. This emphasizes the crucial role of these CTLs in mounting immunity against the JCV.40 The striking association of disease with HIV infection and its occurrence in ICL in the absence of HIV infection suggest that CD4 + T-cells also play an important role against JCV.41 Conversely, humoral immunity is not protective against JCV infection.
The importance of intact host immunity is clear from both the context of PML in patients with immune defects and its remission after HAART.42,43 Remission of the disease is often associated with re-establishment of CD4+ cells and CTLs in the blood and CSF.44–46
Clinicopathologic Syndromes of CNS JCV infection
Until recently, PML was the only known manifestation of CNS JCV infection with nonspecific clinical presentations but typical histopathologic and imaging findings. With restoration of immunity with HAART, there may be an abrupt change in the clinical behavior and histopathologic and imaging manifestations of PML in some patients. This altered presentation has profound clinical significance and needs to be differentiated from the classic presentation. Also 3 new CNS manifestations of JCV infection have been recently recognized and described. For better understanding, all the manifestations of JCV infection are classified below (Table 1).
JCV-associated CNS diseases
cPML
Clinical presentations of PML are nonspecific. In approximately 25% of patients, PML is the initial AIDS-defining illness.47 The cPML presentation begins with focal neurologic deficits that depend on the location of the lesions. Most commonly, patients present with hemiparesis or hemisensory defects. There may be visual problems if there is occipital lobe or optic radiation involvement, language problems if there is involvement of the dominant parietal lobe, and ataxia or dysmetria if there is cerebellar involvement and so forth.35 Usually initial symptoms are partial and gradually worsen, depending on the area of brain involved as lesions enlarge. Approximately 20% of patients develop seizures in addition to the focal neurologic symptoms.48 Patients may also present with cognitive deficits. Occasionally, it may be very difficult to differentiate PML from HIV encephalopathy on the basis of clinical presentation.
Histopathologically, the principal feature is demyelination. Initial foci of demyelination expand and coalesce into larger areas. In advanced cases, lesions may undergo central cavitary necrosis.38 Characteristic histopathologic findings are lytic infection of the oligodendrocytes, which are swollen with enlarged densely basophilic nuclei filled with eosinophilic inclusion bodies and positive staining of the infected oligodendrocytes/nuclei for JCV proteins and nucleic acids.38 The classic infected oligodendrocytes are seen predominantly at the advancing margin of the lesion.49 The oligodendrocytes swell at the expense of extracellular space, which explains diffusion restriction on DWI in the active margin of the lesions. JCV also infects the astrocytes, which are also enlarged, containing numerous enlarged processes. These swollen astrocytes also contain JCV protein and/or gene products. Sometimes enlarged astrocytes contain multilobulated hyperchromatic nuclei, resembling neoplastic cells, which pathologists refer to as ‘“bizarre astrocytes.”50 Another characteristic histopathologic finding of PML is mild or absent inflammation.50 Vary rarely, there may be hemorrhage within the lesions.51
iPML
cPML, as stated earlier, is typically characterized by a distinct lack of inflammatory change in the affected brain tissue. Rarely, reactivation of the JCV and development of PML can be associated with a marked inflammatory reaction. These lesions are characterized by either diffuse or focal perivascular mononuclear infiltrates, mostly of CD3 T-cells, monocytes, or macrophages and B-lymphocytes, CD4 T-cells, and plasma cells.52–54 Radiologically, the lesions are characterized by contrast enhancement or/and mass effect with vasogenic edema.
There may be 2 different settings of the iPML. More commonly, iPML develops in the setting of IRIS in HIV-positive patients following treatment with HAART (see “IRIS and NIRIS”). As expected, patients with IRIS-associated iPML generally have worsening of the neurologic symptoms of cPML. iPML may rarely be the presenting phenotype in non-HIV patients52, as well as in HIV-positive patients without HAART. iPML in non-HIV setting has worse prognosis.52
JCVGCN
Posterior fossa involvement is frequent both in cPML and iPML. Posterior fossa lesions typically affect the middle cerebellar peduncles and adjacent pons and/or cerebellar hemispheres. There is another cerebellar manifestation of JCV, the JCVGCN,55 which infects only cerebellar granule cell neurons, sparing the oligodendrocytes. The classic histopathologic appearances of PML with oligodendrocytic and astrocytic changes are, therefore, not present in this condition. Patients present with isolated cerebellar symptoms, including ataxia and dysarthria. Tropism for cerebellar granular cells is believed to be due to a unique mutation of the VP1 gene of the virus.56
JCM
CSF testing for JCV or JCV DNA is not routinely performed in the work-up of a patient presenting with clinical symptoms of viral meningitis. Blake et al57 first described JCV associated with meningoencephalitis in an immunocompetent girl in 1992. They supported their hypothesis with increasing titers of JCV immunoglobulin G and immunoglobulin M. In a large study, JCV DNA was identified from the CSF of 2 of the 89 patients (19 HIV-positive and 70 HIV-negative) being evaluated for meningitis.58 Both the JCV-positive patients were from HIV-negative group. The authors concluded that tests for BK virus and JCV should be included in the investigative program for patients with meningitis or encephalitis. Viallard et al59 reported a patient with a long history of SLE who presented with acute meningitis with no history of encephalitis or PML. In an extensive work-up of the patient, the authors found that JCV was the only pathogen identified in the CSF. The authors concluded that if CNS infection is suspected in patients with SLE, JCV infection should be considered in the differential diagnoses. CSF PCR for JCV DNA should be rapidly performed to initiate prompt antiviral therapy.
JCE
JCE60 is a newly described encephalopathic form of CNS infection by JCV. Wüthrich et al60 reported a patient with abnormality of higher CNS functions with no focal neurologic deficit. On histology, there was preferential infection of the cortical pyramidal neurons and astrocytes located in the cortical gray matter and gray-white junction with areas of necrosis. The authors found extensive infection of the pyramidal cell neurons and confirmed JCV proteins in the nuclei, axons, and dendrites of pyramidal cell neurons by using double immunostaining methods. Although there was white matter involvement on MR imaging in the late stage, there was only meager infection of the oligodendrocytes without the “typical” demyelination found in PML.
IRIS and NIRIS
IRIS is paradoxic deterioration of clinical response encountered in HIV-infected patients who have received HAART. The diagnosis is often challenging, treatment options are limited, and the prognosis is variable.61 The diagnosis of IRIS is advocated when a patient meets the following criteria62: known HIV-positive patient receiving HAART with a decrease in the HIV-1 ribonucleic acid level from baseline and an increase in CD4+ cells from baseline with clinical symptoms consistent with an inflammatory process rather than an expected course of previously diagnosed opportunistic infection or the expected course of newly diagnosed opportunistic infection or drug toxicity. In the CNS context, the disease may be called NIRIS.
Patients who are antiretroviral naïve are particularly at risk for IRIS.63,64 Other risk factors include the duration and extent of immunodeficiency, polymorphisms in cytokine genes,65 high initial viral load, and the velocity of immune reconstitution.66 No difference in the risk of developing IRIS has been observed when comparing different drug regimens.63 However, initiation of HAART soon after the diagnosis of opportunistic infection may be a clinical predictor of IRIS.67
IRIS may occur during either of the 2 phases of immune restitution that occur after the initiation of HAART.68 The first period of susceptibility occurs in the initial weeks when the increase in CD4 T-cells is largely due to the redistribution of pre-existing memory T-cells. The late phase is a direct result of the proliferation of naïve T-cells, usually after 4–6 weeks but can be as long as 4 years after the initiation of HAART.69
HAART-induced restoration of a pathogen-specific immune response contributes to the pathologic recognition of JCV antigens either in already manifested PML (phase 3 of the pathogenesis) or in the persistent infection phase (phase 2 of the pathogenesis) of PML.70 Although histopathologic criteria have not yet been defined, IRIS is often dominated by CD8+ T-lymphocyte inflammatory infiltrates. Mycobacterial infections are the most common IRIS-associated infection elsewhere in the body.71,72 However, in the CNS, the most common inciting agent is JCV.42,72 Less common IRIS-associated pathogens are Cryptococcus,73,74 herpes virus, and cytomegalovirus.68 Rarely, autoimmune diseases and neoplasia may also incite IRIS.73
PML-IRIS accounts for as many as 18% of the HIV-infected patients with PML.75 As noted previously, PML and IRIS may develop simultaneously in neurologically healthy patients with the start of HAART (unmasking IRIS) or there may be worsening neurologic symptoms in patients with previously manifested PML due to development of IRIS following initiation of HAART (paradoxic IRIS).76 Although there is no demographic difference between these 2 cohorts of patients, the latter group progresses to IRIS during a shorter period compared with the first group, probably due to greater lesion loads.77 Most PML-IRIS cases are characterized by mild symptoms and limited CNS inflammation.
The typical histopathologic appearance of PML-IRIS is hypercellular gray and white matter with gliosis, atypical hyperchromatic astrocytic nuclei, macrophages, and moderate perivascular inflammation, which explains enhancement on contrast-enhanced MR imaging, unlike cPML. While contrast enhancement may be considered as a surrogate marker for the development PML-IRIS, it is present in only 56% of patients.77 Therefore, nonenhancement of a PML lesion with clinical deterioration does not preclude the diagnosis. Unfortunately, to date, there is no biomarker to confirm development of IRIS.
Ironically, PML-IRIS is treated with steroids and a transient antiretroviral drug holiday. PML-IRIS has a favorable outcome if treated appropriately with steroids.77
Diagnosis
Early diagnosis of PML or other JCV-associated CNS infection is of profound importance due to recent expansion of populations of individuals at risk for JCV infection. Even though highly sensitive tests for JCV DNA detection are available and there are specific imaging findings, brain biopsy with histopathologic examination is the criterion standard for the diagnosis of PML. Classic histopathologic changes of cPML and iPML have been described in previous sections. Sensitivity and specificity of brain biopsy are 64%–96% and 100%,78 respectively, with estimated associated procedural complication in 2.9% and morbidity in 8.4%.79
If brain biopsy is not an option, as with debilitated or unwilling patients or inaccessible lesions, diagnosis of PML can be established by brain imaging or demonstration of JCV DNA by PCR of the CSF. Before the introduction of HAART, PCR for JCV DNA was very sensitive and specific with 72%–92% sensitivity and 92%–100% specificity for the diagnosis of PML.80 Recently, however, it has become common to have negative PCR results in patients with AIDS with clinical and imaging presentations indistinguishable from those of PML. It may be possible that the immune restoration with antiretroviral therapy is associated with decreased viral replication and increased clearance of JCV DNA from the CSF.42 As a result of this, the sensitivity of PCR testing for JCV DNA has dropped to 58%.81,82
Imaging has become very important in the diagnosis of PML in the post-HAART era. In fact, most recently published diagnostic criteria classify PML as “definite PML” or “presumptive PML” on the basis of clinical presentation and imaging appearances with or without positive brain biopsy/PCR (Table 2).38 Diagnosis of “definite PML” is considered when there are clinical and imaging findings consistent with PML plus evidence of JCV DNA in the CSF or presence of typical histopathologic changes with demonstration of JCV DNA/protein in the infected cell by in situ techniques (Table 2). The diagnosis of “presumptive PML” is considered when there is evidence of typical imaging and clinical findings with no documentation of JCV (either brain biopsy or lumbar puncture was not performed or JCV DNA was not detected in the CSF).
Diagnostic criteria of PML
The diagnosis of JCVGCN and JCE is confirmed by demonstration of JCV DNA/protein in the infected neuron by using double immunostaining methods.
Imaging
Imaging plays a pivotal role in diagnosis and follow-up of JCV infection. Physicians and patients alike are often reluctant to proceed with invasive brain biopsy. Furthermore, HAART reduces the diagnostic sensitivity of CSF PCR. Given the positive impact of treatment on survival times in these patients, understanding and recognizing the radiologic spectrum are important.
cPML
In the late 1980s, there were a few scattered case reports and small case series introducing MR imaging and CT features of PML.83–86 The classic CT finding was focal (or multifocal) nonenhancing white matter hypoattenuation without mass effect. Posterior fossa lesions cannot be evaluated properly by CT scan due to artifacts.
MR Imaging
MR imaging is the technique of choice for evaluation of PML.87 The imaging findings on conventional MR imaging are organized in regard to lesion distribution and characterization.
Lesion Distribution
Whiteman et al49 first described neuroimaging features of PML in a systematic way with clinical and pathologic correlation in 1993. Typically, PML is a confluent, bilateral but asymmetric, supratentorial white matter disease. However, it can be unilateral, and there may be a single lesion.49,88 CNS involvement can be categorized in the following manner.
White Matter Lesions
Supratentorial.
Because JCV has tropism to oligodendrocytes, any area of the brain may be affected. Asymmetric multifocal bilateral confluent supratentorial lobar white matter involvement is the most common manifestation.49,88 However, there may be only 1 lesion restricted to subcortical U-fibers,89,90 and this may be mistaken for stroke.91 The parietal lobe is most commonly involved, followed by the frontal lobe. Supratentorial lesions typically involve subcortical white matter with a scalloped appearance.92 Centrum semiovale and periventricular white matter can also be involved. White matter involvement has been reported to start in the subcortical regions, the site of highest blood flow, and then to move into the deeper white matter in the centrum semiovale and periventricular regions.93 Internal capsule, external capsule, and corpus callosum involvement are less common. Figure 1 demonstrates an example of a typical supratentorial cPML lesion.
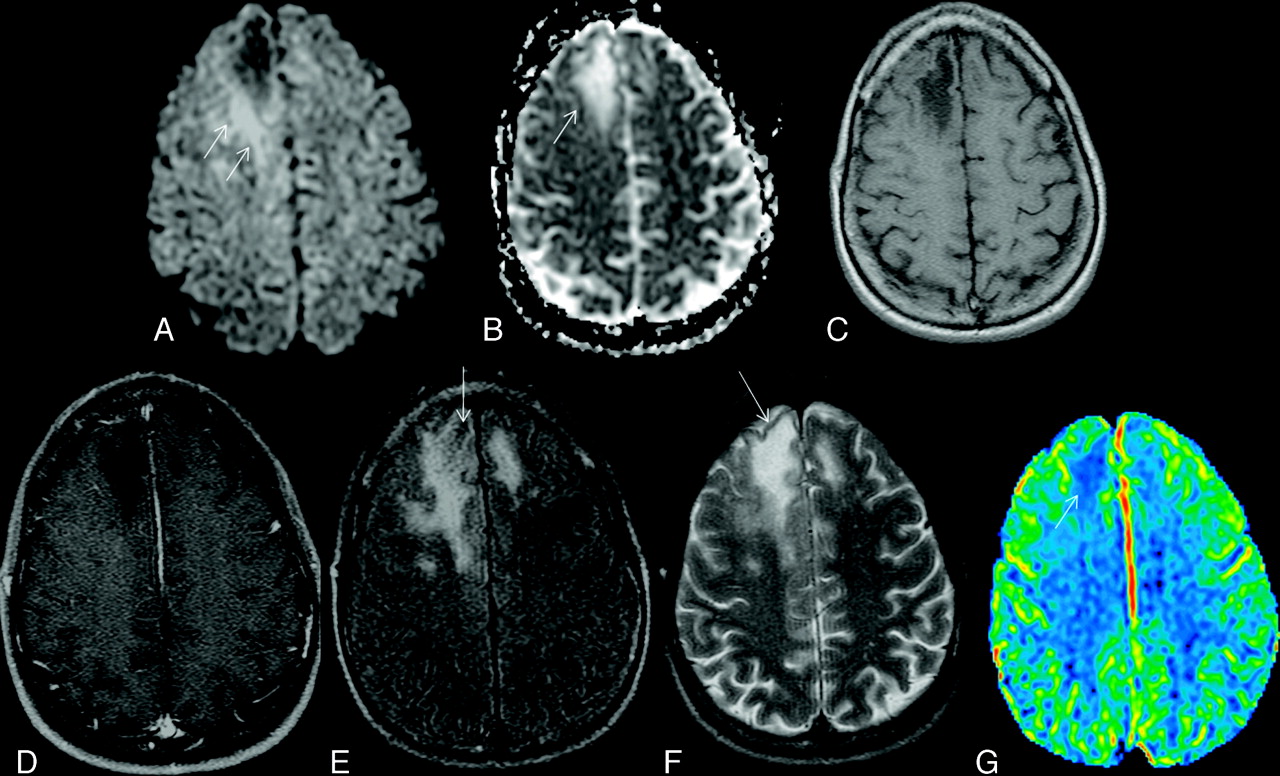
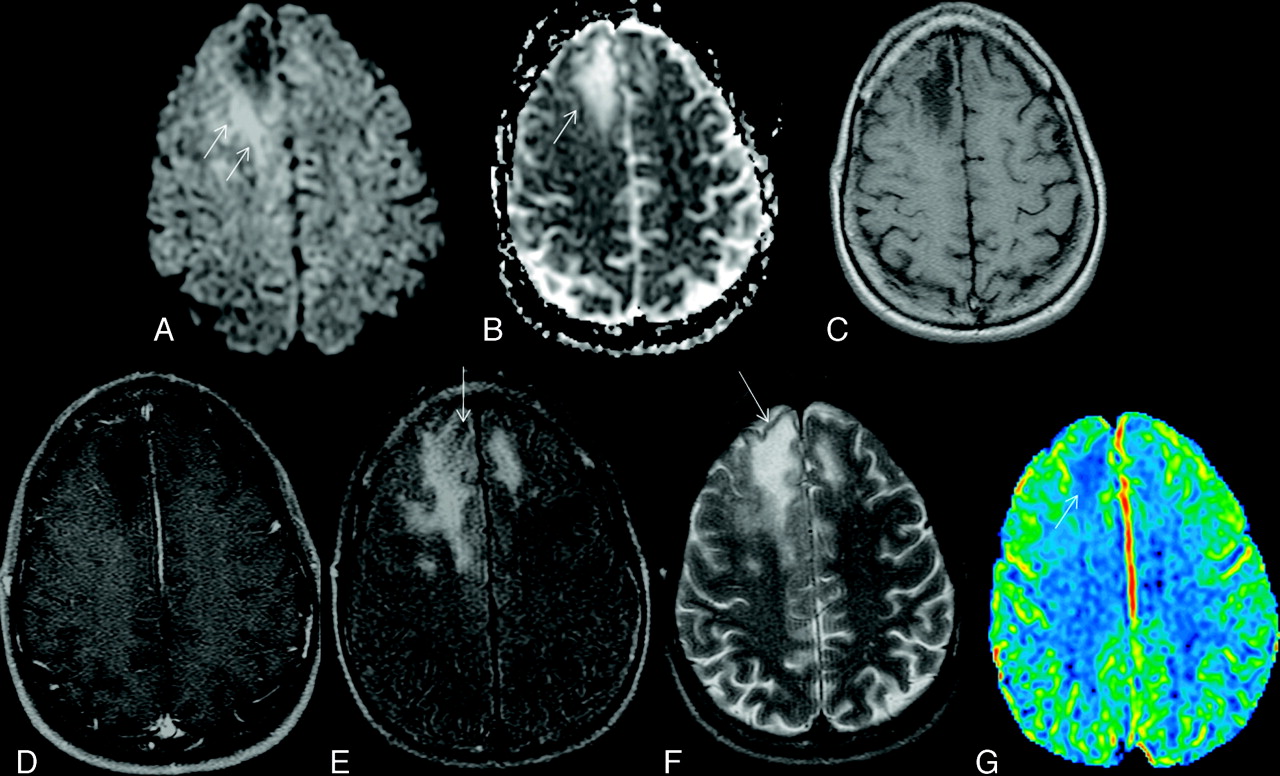
Typical supratentorial right frontal cPML in an HIV-positive patient. A, On DWI, the lesion shows typical restricted diffusion along the advancing edge posteriorly (arrows) and facilitated diffusion centrally. B, On the ADC map, ADC value is low at the posterior advancing edge (arrow) and high at the center of the lesion. C, The lesion typically involves the subcortical U-fiber and is hypointense (relative to gray matter) on the T1-weighted sequence. Note the absence of mass effect from this moderate-sized lesion. D, There is no enhancement of the lesion on the postcontrast T1-weighted sequence. E, On the FLAIR sequence, there is hyperintensity in most parts of the lesion. Note the inversion of the FLAIR high signal anteriorly (arrow) due to intralesional cystic change. F, On the T2-weighted sequence, the entire lesion is hyperintense. Note the adjacent anterior cortex is relatively spared (arrow). G, On perfusion imaging, cerebral blood volume of the lesion is lower (arrow) than that of the contralateral white matter.
Infratentorial.
White matter of the posterior fossa is the next most common area of involvement.49,88 Typically the disease involves the middle cerebellar peduncle and adjacent pons and cerebellum. In an internal case review at our institution, all 9 patients with posterior fossa involvement had predominantly middle cerebellar peduncle involvement associated with adjacent cerebellum and/or pontine involvement. Pontine lesions may extend to the midbrain and/or medulla. Isolated cerebellar white matter or isolated medullary involvement is less common. Figure 2 demonstrates a typical infratentorial cPML lesion.

Typical infratentorial cPML in another HIV-positive patient classically involving the middle cerebellar peduncle (left). A and B, This lesion also has diffusion restriction at the anterolateral advancing edge (high DWI signal intensity and low ADC value, arrows) and diffusion facilitation at the center as evidenced by the DWI (A) and the ADC map (B). C and D, The lesion shows typical hypointensity on the T1-weighted sequence (C) and hyperintensity on FLAIR (D). Note, there are no mass effect. There is no enhancement in the postcontrast T1-weighted sequence (E).
Spinal Cord.
Spinal cord involvement in PML is exceedingly rare. There are only a handful of autopsy reports of the spinal cord PML.94,95 As in the brain, spinal cord PML is typically limited to white matter tracts. Takeda et al94 described involvement of lateral and anterior columns of all 26 spinal segments in a patient with lymphocytopenia. To date, to our knowledge, there is no reported imaging of spinal cord PML.
Gray Matter Lesions
PML may involve gray matter as well. The thalamus is the most common area, followed by the basal ganglia.49,88 Usually gray matter lesions are associated with white matter involvement in almost all cases. Vary rarely, PML lesions can be isolated to the gray matter.96,97
Lesion Location
In the supratentorial type, lesions are typically limited to subcortical U-fibers surrounded by uninvolved cortical tissue.93 Confinement within subcortical U-fiber regions is considered to be a characteristic finding of PML and is used to differentiate PML lesions from HIV encephalopathy and other white matter diseases.93 Unlike other white matter diseases, PML lesions usually spare periventricular or deep white matter. However, with disease progression, adjacent white matter may be involved as well.
In the infratentorial type, lesions are classically located in the middle cerebellar peduncles, frequently extending to adjacent pons and/or cerebellum. In advanced disease, lesions can extend to the midbrain above and the medulla below.
Imaging Appearances
PML lesions are characteristically hypointense on T1. This low T1 signal intensity is considered a differentiating feature from HIV encephalopathy.98 With the initiation of HAART and with disease progression, there is an even further dramatic drop of T1 signal intensity.82,86 Less commonly, lesions may be T1 isointense.88 In some of the cPML lesions, there may be an incomplete hyperintense rim on precontrast T1-weighted sequences (as demonstrated in Fig 3) at the advancing edge. We saw this sign in 4 of our 15 patients. The exact histopathologic correlate of this has not been established. Cinque et al38 described the frequent presence of foamy (lipid laden) macrophages, in response to myelin breakdown. T1 hyperintensity in the advancing edge of some cPML lesions may be attributed to the presence of these macrophages (Figs 1 and 6). Because the presence of little or no inflammation is the classic histopathologic finding of PML, there is no breach of blood-brain barrier and the lesions typically do not enhance.

There is an incomplete rim of T1 hyperintensity (arrows) at the advancing edges of the supratentorial (A) and infratentorial (B) cPML lesions.
On T2-weighted sequences, lesions appear hyperintense to the cortex. Hyperintensity is seen within the lesion and also in the adjacent white matter, which is more prominent on FLAIR. On T2-weighted sequences, the demarcation of the lesion margin from the adjacent uninvolved gray matter is clearly seen (Fig 1). If PML progresses, smaller lesions coalesce, as seen in Fig 4. With progression or involution of a lesion, the central area becomes necrotic and appears hyperintense on T2 and is often attenuated on FLAIR sequences (Figs 1 and 6). In some cases, there may be microcysts at the center of an active lesion on T2-weighted sequences (Fig 5). We have seen microcysts in 6 of our 15 patients with cPML.
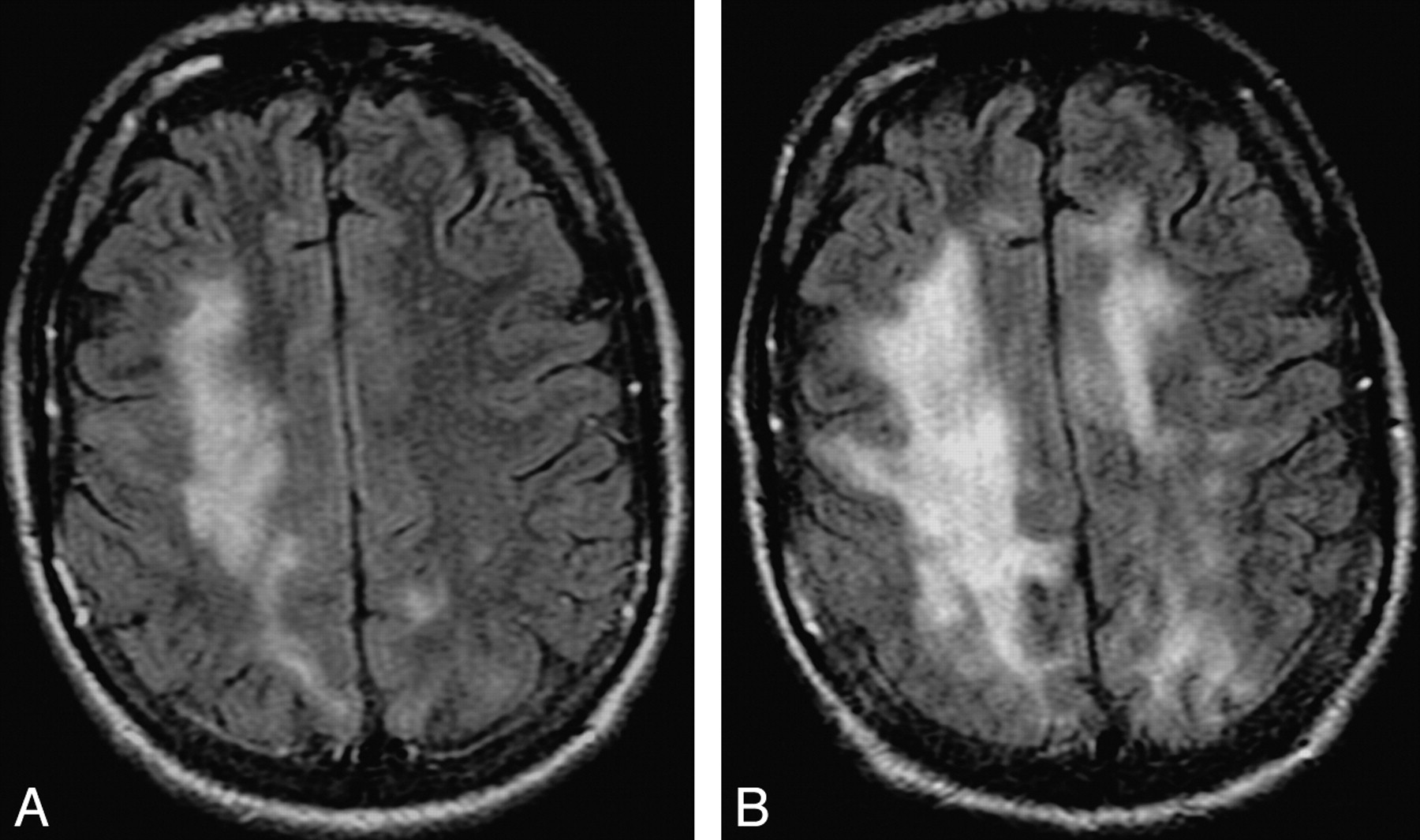
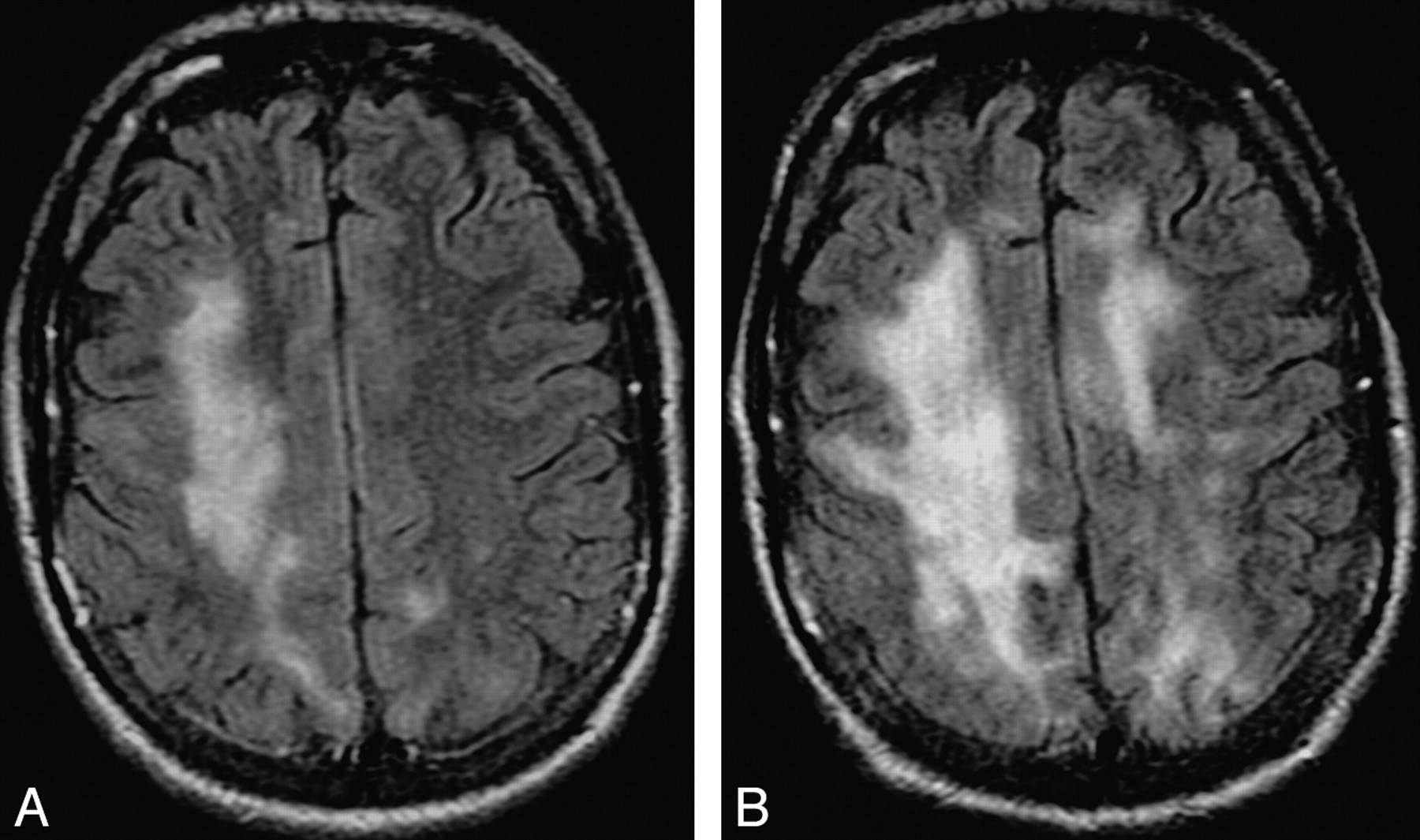
Progression of cPML. A and B, These FLAIR images, 4 months apart, from an HIV-positive patient with cPML demonstrate typical progression of the supratentorial white matter FLAIR hyperintensity.

A, Intralesional microcyst on T2-weighted sequence on the left parietal cPML lesion in an HIV-positive patient, zoomed up in the inset. The arrow points to a microcyst. B, There are 2 intralesional microcysts (arrows) in this right fronto-parietal cPML lesion in another HIV-positive patient.
Another typical imaging finding is absence of atrophy in the active stage.
Imaging of PML in Patients with MS
As briefly mentioned previously, PML can occur in MS patients treated with natalizumab. It is very difficult to differentiate a new PML lesion from an MS plaque in patients treated with natalizumab. A guideline for MR imaging characterization of new PML lesions in patients with MS was proposed by Yousry et al.99 According to this proposed guideline, the diagnosis of confirmed PML is entertained if all of the 3 following criteria are met:
-
Progressive clinical disease.
-
Typical MR imaging findings.
-
Demonstration of JCV DNA in the CSF.
Features favoring PML over MS (as proposed by Yousry et al99) are the following:
-
Diffuse subcortical rather than periventricular white matter involvement; frequent involvement of posterior fossa.
-
Irregular ill-defined infiltrating edge confined to the white matter.
-
Persistent progression of the lesion confined within the white matter tract.
-
No mass effect even in large lesions.
-
Diffuse increased T2 signal intensity; recently involved areas more T2 hyperintense than the old areas.
-
Initially iso- to hypointense with an incremental drop of T1 signal intensity with time; signal intensity never returning to normal.
-
Typically no enhancement, even in large lesions.
Diffusion Imaging
In PML, the appearance on DWI varies according to the disease stage.101 In new active lesions, there is a rim of diffusion restriction at the advancing edge and a central core of facilitated diffusion (Fig 1 A, -B).101,102 The rim is usually incomplete and signifies active infection.38 Histopathologically, this advancing edge correlates with large swollen oligodendrocytes, enlarged “bizarre astrocytes” with numerous large processes, and infiltration of foamy macrophages.38,49,101 This cellular enlargement constricts the extracellular space, the space of maximum Brownian motion of water. Diffusion restriction at the margin can be either due to constricted extracellular space103–105 or enlarged cells per se due to entrapment of water in motion-restricted intracellular space.106 In old “burnt out” lesions after therapy or at the center of a large lesion, there is facilitated diffusion due to disorganized cellular architecture, increased extracellular space secondary to dead oligodendrocytes, macrophage action, and astrocytic reparative responses.101,107
Diffusion tensor imaging also has an important role in the evaluation of PML. Fractional anisotropy, which reflects the organized architecture of the white matter, decreases in PML, suggesting disorganization of the white matter structure. In fact, fractional anisotropy values may decrease in much earlier stages of the disease, when no lesion is evident on conventional and DWI.107 Diffusion tensor imaging parameters also have shown disorganized cellular architecture at the center of a large confluent lesion.108
Magnetization Transfer Imaging
Dousset et al109,110 noted that due to profound demyelination, there is a very low MTR (22%) in PML lesions compared with the normal white matter (47%) of volunteers. The authors also documented an incremental fall of MTR with disease progression, due to increasing demyelination.
Spectroscopy
Proton MR spectroscopy interrogates the chemical milieu of the neuronal microenvironment. In normal MR spectra, there are notable peaks: a prominent NAA peak as a marker for neuron and axon viability, a prominent choline peak (principally due to phosphatidyl choline) due to membrane constituents, and a Cr peak (principally due to creatine phosphate, which plays a role in maintaining energy-dependent systems in brain cells).111 Because the Cr peak remains fairly stable even in the face of disease, it may be used as an internal control.112 An additional peak may be present from lactate, due to inflammation or neuronal mitochondrial dysfunction or related to active anaerobic glycolytic metabolism.113 If a short TE acquisition is used, it is possible to observe the mIns peak.113 In the brain, mIns is synthesized primarily in glial tissue.114 mIns is considered to be a glial marker, and an increase in its content is believed to represent glial proliferation or an increase in glial cell size. Because both processes may occur in brain inflammation, an increase in mIns may be a surrogate marker for inflammation in the brain.115
In the early stage of HIV infection, when clinical symptoms are absent or mild, NAA/Cr is unchanged (92%–98% of normal value).116–119 With the development of the AIDS complex, NAA/Cr decreases (62%–84% of normal value).116–119 The metabolic abnormalities increase proportionally with the severity of the disease.119
In PML, there is substantial decrease of the NAA peak to the contralateral NAA value, whether measured in relation to Cr or by absolute quantification.120 This may signify neuronal loss in the PML lesions.113 The choline peak is elevated, perhaps reflecting myelin destruction.113,120 The mIns peak may be normal or significantly elevated compared with the contralateral normal-appearing white matter. The level of mIns depends upon the stage of the disease. In early and active stage, there is increased level of mIns that gradually decreases to normal level in late quiescent stage.120 Elevation of the mIns/Cr ratio, which signifies local glial-only proliferation secondary to inflammation, has recently been described as a prognostic marker of the disease. In acute lesions, a significantly increased mIns/Cr ratio is associated with increased survival time probably due to more intense inflammation that hinders PML disease progression.115
With this explanation, it is possible that the mIns peak may also be elevated in PML-IRIS because of the associated intense inflammation. To date, to our knowledge, there is no available literature on the spectroscopic appearance of PML-IRIS.
Perfusion Imaging
Histopathologically, PML lesions are not vascular. We obtained perfusion imaging only in 2 patients with cPML. As expected, in both patients, cPML lesions showed lower cerebral blood volume compared to the contralateral normal appearing white matter (Fig 1).
Angiography
Most PML lesions are angiographically negative because there is little or no inflammation. However, Nelson et al121 described pathologic parenchymal blush and arteriovenous shunting in 4 of the 6 patients of their series, which on histopathology turned out to be due to increased small vessel density and “robust inflammatory” changes. The authors concluded that this increased microvascular density was due to neoangiogenesis versus accelerated contrast passage secondary to altered microvascular tone in the presence of different inflammatory kinins. The latter mechanism is a more likely explanation for the angiographic abnormality because all 4 patients had features of iPML on cross-sectional imaging (ie, either mass effect or contrast enhancement).
Nuclear Imaging
Because cPML lesions are pathologically neither inflammatory nor neoplastic, they are not identified in the nuclear medicine studies. In a study of 6 patients, Iranzo et al122 reported no uptake in any of the patients with positive MR imaging signs. In the study of Lee et al,123 1 of the 3 patients with PML did not show any uptake either in the thallium 201 or the gallium 67 scan. However, in this study, 2 of the 3 patients showed both thallium 201 and gallium 67 intake. Although MR imaging or histopathologic features of these 2 patients were not mentioned in the article, probably these 2 patients had iPML. Port et al124 described a patient with PML with uptake of thallium 201, contrast enhancement on the MR imaging, and numerous macrophage infiltrations on histopathology. Although the term “iPML” was not introduced at that time, the authors concluded that the uptake of thallium 201 in that patient was due to an “inflammatory reaction.”
Theoretically, all cPML should have negative nuclear scintigraphy findings, and all iPML lesions should have positive nuclear scintigraphy findings. A systematic study is needed to prove this hypothesis.
Signs of Disease Progression
Increasing confluence and extent of white matter lesions, increasing cortical atrophy, and incremental drop of T1 signal intensity signify disease progression and poor prognosis.88 Figure 6 demonstrates the typical progression of the disease with time even with use of HAART. In the late stage of the disease, there is generalized atrophy and diffuse white matter involvement (Fig 7). Associated HIV encephalopathy may augment the process.

Typical disease course of cPML in an HIV-positive patient receiving HAART. Top panel, a set of images at presentation with focal diffusion restriction (A and B) and very subtle but typical hypointensity on T1 (C) and hyperintensity on FLAIR (D). This initial study was confused with acute subcortical infarction. Middle panel, a set of images 1 month after the initial presentation. No HAART was administered before this scanning. Now the lesion has enlarged in size with typical diffusion restriction (arrows) at the medial and posterior advancing edges (E and F). Now the T1 hypointensity is more obvious (G). The adjacent cortex is not involved (arrows). Typically the lesion is hyperintense on FLAIR (H). Bottom panel, a set of images 19 months after initial presentation. The patient received HAART for 18 months. Now there is no diffusion restriction (I). On the T1-weighted sequence (J), there is profound T1 hypointensity associated with new/progressive atrophy. There is FLAIR hyperintensity in the adjacent areas. However, the main lesion is not hyperintense on FLAIR (K). On T2 (L), the lesion itself is very hyperintense compared with the adjacent white matter, suggesting cystic encephalomalacia. Note that the adjacent cortical architecture is preserved.

Sequel of cPML. A and B, The 2 T2-weighted sequences, 16 months apart, from an HIV-positive patient demonstrate prominent brain atrophy with dilation of the ventricles and prominence of the sulci. Even at this advanced stage, there is minimal cortical involvement.
Can Imaging Monitor Treatment?
According to Thurnher et al,92 differentiation between treatment responders versus nonresponders can be made with FLAIR signal intensity; findings of progressively decreasing T1 and FLAIR signal intensity on follow-up imaging indicate the burnt out part of the PML lesions due to leukomalacia and associated atrophy (bottom panel of Fig 6). On the other hand, increasing FLAIR intensity and progressive T1 hypointensity indicate progressive disease and are poor prognostic signs.109 In an isolated case report, Usiskin et al108 documented restoration of white matter anisotropy in a pathologically proved PML case in response to HAART by using high-b-value DWI.
Can Imaging Predict Prognosis?
In a pathologically proved large series, Post et al88 found no correlation between lesion size, lesion location, signal intensity, brain atrophy, or hydrocephalus and patient survival. However, there was significant positive correlation between risk of death and mass effect at baseline imaging. Also there was a 2-fold increase in risk of death in patients with basal ganglia gray matter involvement. In this study, there was a trend toward increasing survival in patients with multiple discrete lesions versus large confluent lesions. In a comparative study between short-term survivors and long-term survivors, Thurnher et al92 noticed that extensive brain involvement was associated with longer survival time. Increased mIns/Cr ratio in the lesion also signifies favorable prognosis.115
iPML
The imaging manifestation of iPML is exactly like that of cPML except that the lesion has additional peripheral enhancement and/or mass effect due to inflammation. Rarely, the contrast enhancement is so subtle that it may not be identified on a regular spin-echo T1 sequence and can be detected only by using a magnetization transfer imaging pulse on T1 sequences. Due to an inflammatory infiltrate, these lesions may have high intake in nuclear scintigraphy scanning.
JCVGCN
In JCVGCN,55 there is isolated involvement of the internal granular cell layer of the cerebellum without white matter involvement. In the early stage of the disease, there is no specific MR finding. In later stages, there is isolated cerebellar atrophy followed by increased T2 signal intensity.
JCM
There is no specific MR imaging finding of JCM.
JCE
Unlike PML, lesions in JCE are initially restricted to hemispheric gray matter with extension to the subcortical white matter with progression of the disease. Like cPML, lesions do not enhance on contrast.58
Treatment
There is no specific treatment for JCV infection. In HIV-infected patients, optimization of HAART is the best therapeutic choice. HAART may stabilize the clinical and imaging manifestations of the disease in ≤50%-60% of HIV-positive patients with PML.38 In HIV-negative patients, removal of the cause of immunosuppression (steroids, calcineurin inhibitors in transplant patients, natalizumab etc) as much as clinically allowable is the treatment of choice.125 A number of specific drugs against JCV such as cytarabine, cidofovir, and topotecan were used in multiple clinical trials. All these investigational drugs showed either no clinical benefit or possible benefit at the expense of high toxicity.38 In patients with PML-IRIS, steroid should be started in patients with inflammation-induced worsening of symptoms or signs.38
Conclusions
It is now clear that the term “PML” fails in defining the disease spectrum of JCV infection. JCV infection may not have a progressive course, may not be multifocal, and may not be contained within the white matter. The appropriate terminology should be based on histopathology and/or imaging appearances. The term “PML” alone should be avoided in favor of the more descriptive terms cPML or iPML, on the basis of the imaging and or histopathologic appearances. Also, neurotropic manifestations of JCV infection (JCVGCN, JCE, etc) should not be confused with either type of PML.
Indicates open access to non-subscribers at www.ajnr.org
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Abbreviations
- Epidemiology: Population at Risk
- Pathogenesis
- Clinicopathologic Syndromes of CNS JCV infection
- IRIS and NIRIS
- Diagnosis
- Imaging
- Diffusion Imaging
- Magnetization Transfer Imaging
- Spectroscopy
- Perfusion Imaging
- Angiography
- Nuclear Imaging
- Signs of Disease Progression
- Can Imaging Monitor Treatment?
- Can Imaging Predict Prognosis?
- iPML
- JCVGCN
- JCM
- JCE
- Treatment
- Conclusions
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- Clinical Reasoning: A 71-Year-Old Man With Horizontal Gaze Palsy, Anarthria, and Quadriparesis
- Clinical Reasoning: A 52-year-old woman with 3 weeks of progressive gait ataxia and dysarthria
- Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions
- Punctate pattern: A promising imaging marker for the diagnosis of natalizumab-associated PML
- Brain Magnetic Susceptibility Changes in Patients with Natalizumab-Associated Progressive Multifocal Leukoencephalopathy
- Unusual Case of Progressive Multifocal Leukoencephalopathy After Allogeneic Hematopoietic Stem-Cell Transplantation
- CNS-Immune Reconstitution Inflammatory Syndrome in the Setting of HIV Infection, Part 1: Overview and Discussion of Progressive Multifocal Leukoencephalopathy-Immune Reconstitution Inflammatory Syndrome and Cryptococcal-Immune Reconstitution Inflammatory Syndrome
- Cranial nerve involvement in infratentorial progressive multifocal leukoencephalopathy
- Progressive multifocal leukoencephalopathy in a patient with transitory lymphopenia