
Abstract
BACKGROUND AND PURPOSE: Pituicytoma, SCO, and GCT are poorly understood entities with confusing nomenclature and undetermined imaging characteristics. Our purpose was to confirm published cases of pituicytoma, SCO, and GCT with the newest 2007 World Health Organization criteria and elucidate imaging findings that distinguish these tumors from common entities such as pituitary adenoma.
MATERIALS AND METHODS: A literature search identified 145 published cases (81 GCTs, 48 pituicytomas, and 16 SCOs). Case diagnoses were blindly reviewed by a neuropathologist according to the latest WHO criteria, resulting in 112 pathologically documented cases (64 GCTs, 35 pituicytomas, and 13 SCOs). Imaging illustrations from proved cases were reviewed to determine location, configuration, attenuation and signal intensity, and enhancement characteristics.
RESULTS: Only pituicytomas presented as purely intrasellar lesions (7/33). Most GCTs were purely suprasellar (28/45). All SCOs were both intra- and suprasellar (13/13). Twenty-five percent of pituicytomas (6/22) and GCTs (7/30) appeared separate from the pituitary gland. All SCOs were infiltrating. Seventy-nine percent of entities appeared isointense to brain on T1-weighted image (34/43). Seventy-four percent of pituicytomas enhanced homogeneously (14/19). Twelve of 23 GCTs and 5/7 SCOs enhanced heterogeneously. Most GCTs were hyperattenuated to brain on CT (18/20). Eleven of 13 cases enhanced homogeneously. Visual disturbances were common symptoms for all entities (67/112). Diabetes insipidus was rare (4/112).
CONCLUSIONS: Pituicytoma may be considered for purely intrasellar masses that are clearly separate from the pituitary gland. GCT should receive consideration for purely suprasellar lesions that are hyperattenuated to brain on CT. SCO should be considered for infiltrating pituitary masses with a mixed intra- and suprasellar location. A history of diabetes insipidus helps to exclude these tumors.
ABBREVIATIONS
- DI
- diabetes insipidus
- GCT
- granular cell tumor
- SCO
- spindle cell oncocytoma
Primary nonadenomatous pituitary gland tumors are rare, poorly understood entities with confusing nomenclature. The 2007 WHO Classification of Tumors of the Central Nervous System clarified and redefined criteria for pituicytoma, codifying it as a separate diagnostic entity distinct from GCT of the neurohypophysis.1 The 2007 update also added a new entity—SCO—to the spectrum of nonadenomatous sellar neoplasms.2 The purpose of our study was to apply the new World Health Organization (WHO) criteria to published cases and elucidate imaging findings that might distinguish these rare neoplasms from each other as well as more common lesions such as pituitary adenoma and lymphocytic hypophysitis.
Materials and Methods
A comprehensive literature search of English and non-English studies by using PubMed and Google Scholar was completed between September and October 2010 to identify all previously published cases of pituicytoma, SCO, and neurohypophyseal GCT. To include all names assigned to these tumors over past decades, the following keywords were used in the search: pituicytoma, spindle cell oncocytoma, granular cell tumor, infundibuloma, “choristoma”, granular cell myoblastoma, Abriksossoff tumor, and pilocytic astrocytoma.
Search results were screened by the primary author to include only those tumors involving the sellar and suprasellar region (eg, most GCTs involve extracranial sites such as the tongue and esophagus3; pilocytic astrocytoma—previously used synonymously for what is now termed pituicytoma—typically does not involve the infundibulum or pituitary gland4). This search identified 145 potential cases (81 GCTs, 48 pituicytomas, and 16 SCOs).
Full-text articles of each report were obtained. The radiologic and histologic images from each article were compiled into electronic databases by the primary author. Captions or other identifying text accompanying each image were removed to facilitate blinded case review.
The histologic image data base was reviewed by a board-certified neuropathologist (S.S.C.) who was blinded as to tumor entity and instructed to classify each case, if possible, as pituicytoma, GCT, or SCO in accordance with the latest 2007 WHO criteria. For cases in which provided histologic images were insufficient to meet WHO criteria, all available written descriptions of the study's pathologic diagnoses were provided. This included descriptions of histology, immunohistochemistry, and ultrastructure. After review of this information, the neuropathologist was again asked whether sufficient WHO criteria were met to classify the case definitively. Any cases that could not be assigned to 1 of our 3 entities based on histology, written reports, or both were recorded as unclassifiable and eliminated from further consideration.
The review process resulted in 112 pathologically documented cases in total from 65 articles (64 GCTs,3,5⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓–44 35 pituicytomas4,45⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓–63 and 13 SCOs64⇓⇓⇓⇓⇓–70). Histologic images were of too poor quality to analyze in 7 cases that were received through interlibrary loan (3 GCTs, 2 pituicytomas, and 2 SCOs), and these were marked as unclassifiable along with 33 additional cases. In no case was a tumor reassigned from 1 entity (eg, pituicytoma) to another (eg, SCO or GCT). Most articles were published in pathology, neurosurgery, or other clinically oriented journals. Only 5 articles were published in radiology journals.
Of the 112 pathologically documented cases, only 58 (30 GCTs, 22 pituicytomas, and 6 SCOs) contained CT images, MR images, or both. Original MR images were available for 19 cases of pituicytoma, 7 cases of SCO, and 24 cases of GCT. T1WI was available for 15 pituicytomas, 4 SCOs, and 24 GCTs. T2WI was available for 8 pituicytomas and 17 GCTs. Results of postcontrast scans were available for 19 pituicytomas, 7 SCOs, and 23 GCTs.
Information regarding CT signal intensity characteristics were available for only GCTs (n = 20). Results of CT enhancement patterns were available in 13 of these cases.
Original CT and MR images were compiled into a database and reviewed by a senior neuroradiologist (A.G.O.). Findings including location (suprasellar/infundibulum, intrasellar/pituitary gland, or both), configuration (round or infiltrating and separation from the pituitary gland), attenuation and signal intensity, and enhancement characteristics were tabulated.
Finally, M.F.C. extrapolated the age, sex, and clinical symptoms at presentation from each pathologically documented case.
Results
Location
Pituicytoma was the only tumor that ever presented as a purely intrasellar lesion (7/33) (Table 1). Most pituicytomas were either suprasellar (13/33) or combined intra- and suprasellar lesions (13/33). GCTs were either suprasellar (28/45) or both intra- and suprasellar (17/45). No SCOs were purely intra- or suprasellar; all SCOs were both intra- and suprasellar (13/13).
Anatomic location for cases of pituicytoma, SCO, and GCT
Separation from Pituitary Gland
Only 25% each of pituicytomas (6/22) and GCTs (7/30) could be clearly separated from the pituitary gland. Seventy-five percent of all tumor entities were infiltrating and could not be separated from the underlying pituitary gland (39/52).
Imaging Findings
Signal intensity characteristics and enhancement patterns from CT imaging were available for only the GCTs. Eighteen of 20 were hyperattenuated compared with brain on noncontrast CT. Postcontrast scans were available in 13/20 cases. Eighty-four percent (11/13) enhanced homogeneously. One tumor (8%) enhanced inhomogeneously and 1 tumor showed no enhancement after contrast administration.
Forty-three cases had 1 or more MR images illustrated (Table 2). T1WI was available in all 43 cases. Seventy-nine percent of all entities, regardless of tumor type, appeared isointense compared with cortex on T1WI (34/43). T2WI was available in only 25 cases. Pituicytomas were generally hyperintense on T2WI compared with gray matter (6/8), whereas GCTs were predominately isointense (10/17). No confirmed cases of SCO had published images of T2-weighted scans.
MR signal characteristics for pituicytoma, SCO, and GCT
Contrast-enhanced T1WI was available for review in 49 cases. Seventy-four percent of pituicytomas enhanced homogeneously (14/19); the remainder showed heterogeneous enhancement (5/19). Enhancement patterns for the other 2 entities were mixed (12/23 GCTs and 5/7 SCOs demonstrated heterogeneous enhancement, whereas the remainder enhanced homogeneously).
Clinical Findings
Visual disturbances (eg, bitemporal hemianopsia, decreased visual acuity) were the most common presenting symptom for all entities (67/112; Table 3). With pituicytomas, the next most common presenting symptoms were headache (10/35), hypopituitarism (9/35), fatigue (8/35), and decreased libido (7/35). For SCO, hypopituitarism was the next most common symptom (6/13) followed by headache (4/13). For GCT, the next most common symptoms were headache (21/64), amenorrhea (12/64), fatigue (8/64), and memory loss (7/64).
Presenting symptoms for pituicytoma, SCO, and GCT
DI was especially uncommon with these tumors. Only 1 pathologically documented case of pituicytoma56 and 3 cases of GCT36,39,44 had DI. Similarly, increased levels of prolactin were reported for only 1 case of pituicytoma,55 2 cases of SCO,64,70 and 4 cases of GCT.11,17,22,31 Galactorrhea was reported only in 1 case of GCT.11
Sex
The male:female ratio was 18:17 (51.4% male) for pituicytoma, 5:8 (61.5% female) for SCO, and 22:42 (65.5% female) for GCT.
Age
The average age at diagnosis was 50.3 years for pituicytoma, 59.4 years for SCO, and 49.2 years for GCT.
Discussion
Pituicytoma, SCO, and GCT of the neurohypophysis are rare, poorly understood neoplasms of the sellar region. Pituicytoma has an especially notable history of frequently changing nomenclature and shifting diagnostic criteria.62 Synonyms for pituicytoma over past decades have included choristoma, granular cell myoblastoma, infundibuloma, pilocytic astrocytoma, and even granular cell tumor. Not until the 2007 edition of the WHO Classification of Tumors of the Central Nervous System was pituicytoma designated as a distinct diagnostic entity, separate from GCT. The 2007 edition also added SCO as a new distinct entity in the differential diagnosis of sellar neoplasms.
Given their striking similarities on imaging to other much more common lesions such as pituitary adenoma, the preoperative diagnosis of pituicytoma, SCO, and GCT has been problematic. The inability to distinguish these lesions from entities such as pituitary adenoma is important because these tumors, unlike pituitary adenomas, tend to be very vascular and are prone to heavy bleeding during surgical resection.5,52,65 This has often resulted in the need to stabilize the patient, abort the surgery, and consider reoperation at a later date, potentially after embolization of tumor vasculature.62 If reoperation does not occur, symptomatic recurrence is common.8
Detecting clues that might establish the preoperative diagnosis of these entities has been problematic due to the exceptional rarity of these tumors. No single institution is likely to see more than a handful of these tumors over the course of several decades. The literature consists mostly of single case reports and a few very small case series. Therefore, only a retrospective meta-analysis of all published cases could potentially detect meaningful differential diagnostic information.
In addition, considering these entities' history of frequently changing and often overlapping nomenclature, confirmation of published case diagnoses by a board-certified neuropathologist by using the new 2007 WHO criteria was a necessary prerequisite to imaging analysis. Confirming the precise histologic diagnosis in each case has allowed us to determine that all cases included in our meta-analysis do indeed represent true cases of pituicytoma, SCO, or GCT.
Major limitations of our study include dependence on the often-limited information provided for the figures, images, and text as included in each original case report or case series. Less than 10% of cases were published in the radiology literature. Precise delineation of imaging parameters were universally absent from nonradiology journals and were present in only 2 of the 5 radiology manuscripts used in our imaging analysis. We did not have access to any of the original patient scans for review or comparison to published reports.
Pituicytoma
The definitive histologic description of pituicytoma46 and the subsequent acceptance of pituicytoma as a distinct tumor entity were only recently established. Given the lack of known clinical and imaging findings specific to pituicytoma, these tumors are typically diagnosed preoperatively as pituitary adenoma.47 Attempted resection of these presumed adenomas often results in unexpected heavy intraoperative bleeding, subtotal resection, and a high risk of symptomatic tumor recurrence.62
Our study identifies specific imaging findings (Fig 1) that could permit a neuroradiologist to make the preoperative diagnosis of pituicytoma in selected cases. Although a minority of pituicytomas will present in this manner, the diagnosis might be suggested if imaging shows a mass that is purely intrasellar and clearly separate from the pituitary gland. We found no similar presentation in any of our pathologically documented cases of SCO or GCT. Such a presentation also would be rare for pituitary adenoma, lymphocytic hypophysitis, or physiologic pituitary hyperplasia.71

Pituicytoma. Sagittal T1WI image (A) and coronal T1WI postcontrast scan (B) show a rounded suprasellar mass that is clearly separate from the pituitary gland. (From Gibbs WN, Monuki ES, Linskey ME et al. Pituicytoma: diagnostic features on selective carotid angiography and MR imaging. AJNR Am J Neuroradiol 2006:27:1639–42. Used with permission.)
Patients with pituicytoma almost never present with diabetes insipidus, galactorrhea, or prolactinemia. When these symptoms are present, more common diagnoses such as pituitary adenoma or lymphocytic hypophysitis are likely. Instead, to support the diagnosis of pituicytoma, the radiologist should look for a classic history of visual disturbance, headache, or both.
Spindle Cell Oncocytoma
SCO (Fig 2) was first described in 2002 by Roncaroli et al69 in their series of 5 cases. Only 16 total cases have been reported in the literature. Of these, we were able to confirm the histopathology of 13 cases. The remaining 3 cases were marked as unclassifiable due to the poor quality of images received through interlibrary loan.
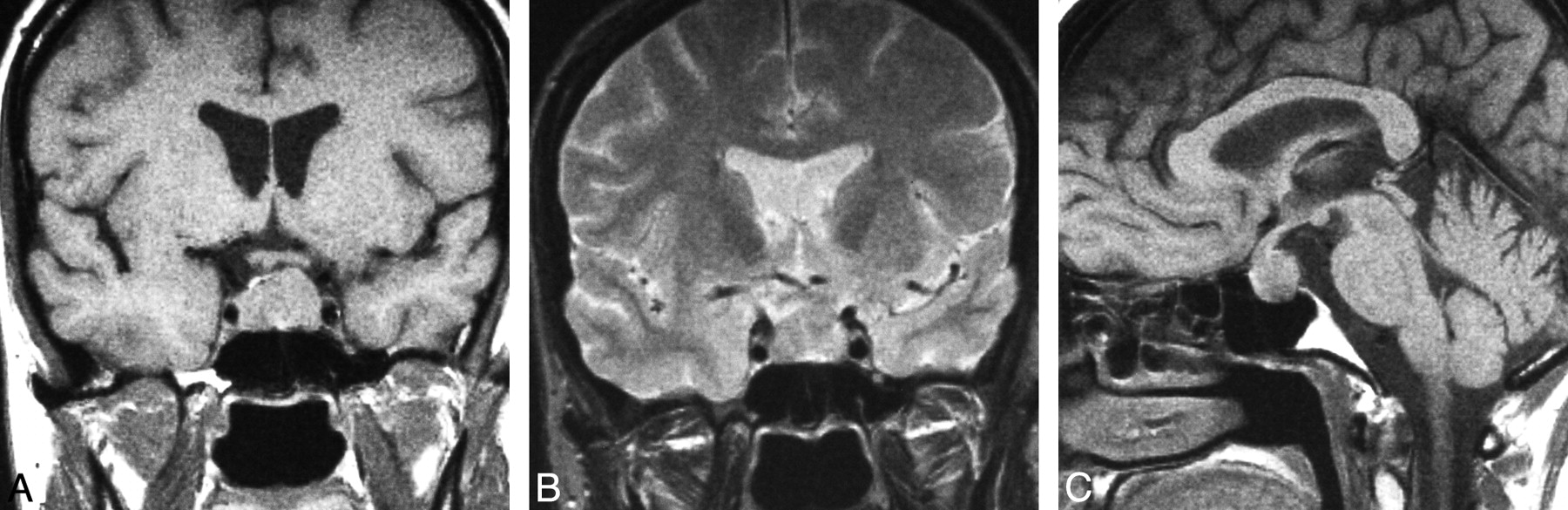
Spindle cell oncocytoma. Coronal T1WI (A) demonstrates a mixed intra- and suprasellar infiltrating pituitary lesion. Coronal T1-postcontrast scan (B) reveals a heterogeneous pattern of enhancement. Sagittal T1WI (C) shows enlargement of the anterior pituitary by the infiltrating mass and displacement of the unaffected neurohypophysis. (From Vajtai I, Sahli R, Kappeler A. Spindle cell oncocytoma of the adenohypophysis: report of a case with a 16-year follow-up. Pathol Res Pract 2006:202:745–50. Used with permission.)
Our analysis of pathologically documented cases of SCO was limited by the small number of imaging studies available in the published literature. However, all 13 cases of the pathologically documented SCO presented as combined intra- and suprasellar lesions. Thus it is unlikely that a lesion presenting as a purely intra- or suprasellar mass on imaging is a SCO.
SCOs arise from the adenohypophysis whereas pituicytoma and GCT derive from the neurohypophysis.69,70 Therefore, if imaging localizes a tumor to the neurohypophysis, the diagnosis of SCO may be excluded. Moreover, all cases of SCO were infiltrating and none could be seen separately from the pituitary gland itself. It is therefore not possible to distinguish SCOs from more common lesions such as pituitary adenoma or lymphocytic hypophysitis on the basis of imaging features alone.
SCO typically presents with visual disturbance, panhypopituitarism, and headache. From our analysis, the incidence of panhypopituitarism seems to be more common with SCO than either pituicytoma or GCT. This discrepancy may possibly be explained by SCO's exclusive derivation from the adenohypophysis. There are no reported cases of SCO presenting with DI.
We could identify no specific imaging or clinical findings that allowed us to suspect the preoperative diagnosis of SCO. In contrast, there are imaging and clinical clues that help exclude SCO when present. These include a mass that clearly arises from the neurohypophysis or masses that are exclusively either intra- or suprasellar on imaging studies. Individuals presenting with a pituitary mass and concomitant diabetes insipidus are unlikely to have a SCO.
Granular Cell Tumor
Boyce and Beadles72 first described GCT (Fig 3) as a distinct neurohypophyseal tumor in 1893. Many GCTs are asymptomatic and consist of small nests of tumor cells that do not have any space-occupying effects.7 Such tumor cell nests are common in the general population. Necropsy of 200 pituitary glands found GCT nests in 5.7% of cases,12 and analysis of 1364 pituitary glands identified GCT nests in 6.45% of cases.73
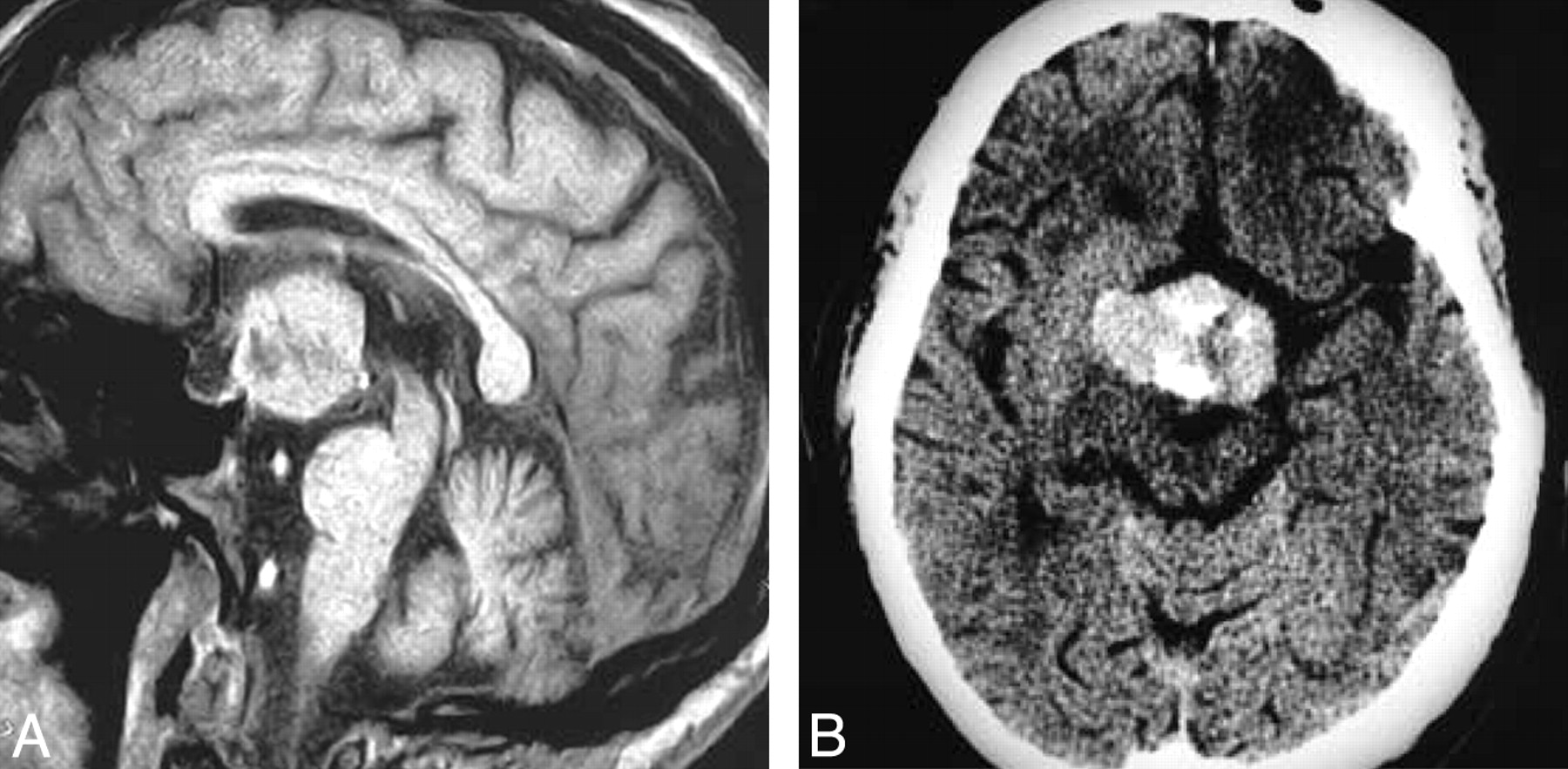
Granular cell tumor. T1-weighted postcontrast scan (A) demonstrates a large suprasellar mass with a heterogeneous pattern of enhancement that is clearly separate from the pituitary gland. Contrast-enhanced axial CT (B) shows a large mass with heterogeneous enhancement that is hyperattenuated compared with brain. (From Buhl R, Hugo HH, Hempelmann RG et al. Granular-cell tumor: a rare suprasellar mass. Neuroradiology 2001:43:309–12. Used with permission.)
As first reported in 1951,42 these tumor cell nests may enlarge over time and become clinically symptomatic, usually in middle-aged or older adults.22 To date, symptomatic GCT of the neurohypophysis has been described in 81 cases, 64 of which we could confirm on histopathologic review.
Our analysis shows that GCT is almost always hyperattenuated to brain on CT and usually demonstrates a homogeneous pattern of enhancement. Most GCTs are entirely suprasellar in location. None of our 64 pathologically documented cases presented as a purely intrasellar mass. Therefore, GCT should be excluded from the differential diagnosis of purely intrasellar lesions.
Clinical findings for GCT are nonspecific and most commonly include visual disturbance, headache, and amenorrhea. Similar to our other entities, DI, prolactinemia, and galactorrhea are uncommon presenting features of GCT.
The diagnosis of GCT should be considered for purely suprasellar masses that are hyperattenuated compared with brain on CT and enhance homogeneously. MR imaging offers few clues that raise preoperative suspicion for GCT. On MR imaging, GCT tends to be isointense on both T1WI and T2WI. The pattern of enhancement for GCTs is equally heterogeneous or homogeneous after gadolinium administration.
Conclusions
Pituicytoma, SCO, and GCT represent rare but important differential diagnostic considerations of sellar and suprasellar lesions. The preoperative diagnosis of these tumors has been difficult owing to a lack of specific imaging and clinical findings. From our analysis, we have identified imaging and clinical clues that make the potential preoperative diagnosis of these entities possible (Fig 4). A diagnosis of pituicytoma may be considered for masses that are purely intrasellar and clearly separate from the pituitary gland on imaging. GCT should receive diagnostic consideration for lesions that are hyperattenuated compared with brain on noncontrast CT and of a purely suprasellar location. SCO should be considered only for infiltrating pituitary masses with a mixed intra- and suprasellar location. Neither pituicytoma, SCO, nor GCT should be highly considered for patients who present with DI, prolactinemia, or galactorrhea. Instead, these rare tumors most often present with visual disturbance and headache.
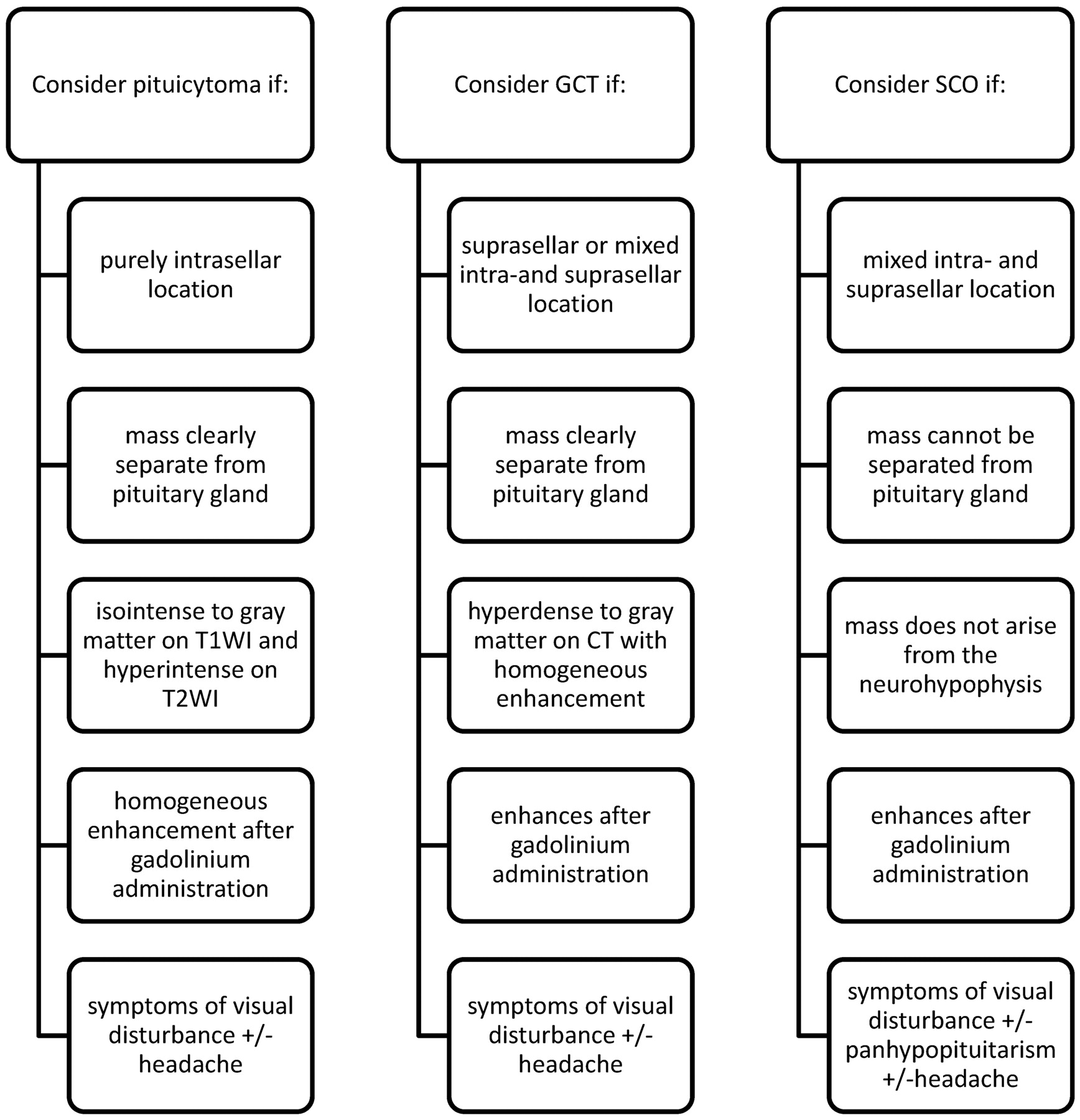
Key diagnostic considerations for pituicytoma, SCO, and GCT.
Footnotes
-
Paper previously presented in part at: Annual Meeting of the American Society of Neuroradiology, June 6–9, 2011; Seattle, Washington.
Disclosures: Anne G. Osborn. Ownership Interest: Amirsys, Amirsys Publishing, Details: shareholder; Other Financial Relationships: Amirsys Publishing, Details: CEO.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- Received February 10, 2011.
- Accepted after revision April 28, 2011.
- © 2011 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}