
Abstract
BACKGROUND AND PURPOSE: Hunter syndrome (MPS type II) is a rare X-linked recessive disease caused by lysosomal enzyme iduronate-2-sulfatase deficiency, characterized by frequent and variable brain and skull involvement. Our objective was determine the frequency of closed cephaloceles in a large cohort of subjects affected with Hunter syndrome and to investigate possible correlations with clinical and neuroradiologic findings.
MATERIALS AND METHODS: Brain MR imaging of 33 patients (32 males and 1 female, age range 2.5–30.8 years, mean age 10.4 years) affected with Hunter syndrome were retrospectively evaluated. Eleven (age range 3.6–30.8 years; mean age 15.1) presented with an “attenuated” phenotype, while 22 (age range 2.5–19.1 years; mean age 8.2) had a “severe” phenotype.
RESULTS: A closed cephalocele was detected in 9/33 patients (27%) at the level of anterior and middle fossa in 6 and 3 cases, respectively; 6/9 subjects were affected with the attenuated phenotype and 1/9 suffered from epilepsy. Closed cephaloceles did not show a significant association with other brain and spine MR imaging features of Hunter disease, such as enlargement of perivascular spaces, cisterna magna, pituitary sella, ventricles and subarachnoid spaces, craniosynostosis, dens hypoplasia, white matter abnormalities, spinal stenosis due to periodontoid cap, platyspondylia, or intervertebral disk anomalies.
CONCLUSIONS: Closed cephaloceles are frequent in Hunter syndrome and should be considered a neuroradiologic feature of this disease.
ABBREVIATIONS:
- ERT
- enzyme replacement therapy
- GAGS
- glycosaminoglycans
- MPS
- Mucopolysaccharidosis
- PVS
- perivascular space
Hunter syndrome (MPS type II, OMIM#309900) is a rare X-linked recessive disease caused by mutations of the gene encoding for iduronate-2-sulfatase, an enzyme involved in lysosomal GAGs catabolism. The accumulation of GAGs in nearly all cell types, tissues, and organs leads to a progressive multisystemic involvement. Clinical manifestations include organomegaly, airway obstruction, cardiomyopathy, joint stiffness, neurologic decline, and typical facial dysmorphism and skeletal deformities.1
Patients are traditionally divided into 2 groups, according to phenotype. The severe form is characterized by clinical onset between 2 and 4 years of age, progressive neurologic involvement, and death in the first 2 decades. In the attenuated form, clinical signs and symptoms occur later, the neurologic deficit is minimal or absent, and patients may survive until late adulthood.2,3
ERT with recombinant human iduronate-2-sulfatase (idursulfase; Elaprase, Shire Human Genetic Therapies, Cambridge, Massachusetts) has been shown to improve many signs and symptoms of the disease.1 However, the true impact on the central nervous system and skeletal and skull abnormalities is still debated.1,4
Several MR imaging findings have been described so far in patients with Hunter syndrome.5⇓⇓⇓⇓⇓–11 In a recent systematic study of brain and spine MR imaging features of patients with Hunter syndrome,12 our attention was drawn by a peculiar herniation of meninx and brain parenchyma, consistent with a meningoencephalocele, in a couple of patients (Fig 1). Meningoencephaloceles are malformations characterized by protrusion of meninges and/or brain tissue due to a skull defect that may be the result of various acquired processes including trauma, surgery, infection, and neoplasm, or of a primitive defect of neural tube closure.13⇓–15 To our knowledge, such a condition has never been previously described in Hunter syndrome; therefore, all the MR imaging examinations were re-evaluated to define the frequency of focal parenchymal and/or meningeal herniation in our cohort and the possible correlation with clinical phenotype or other neuroradiologic abnormalities.

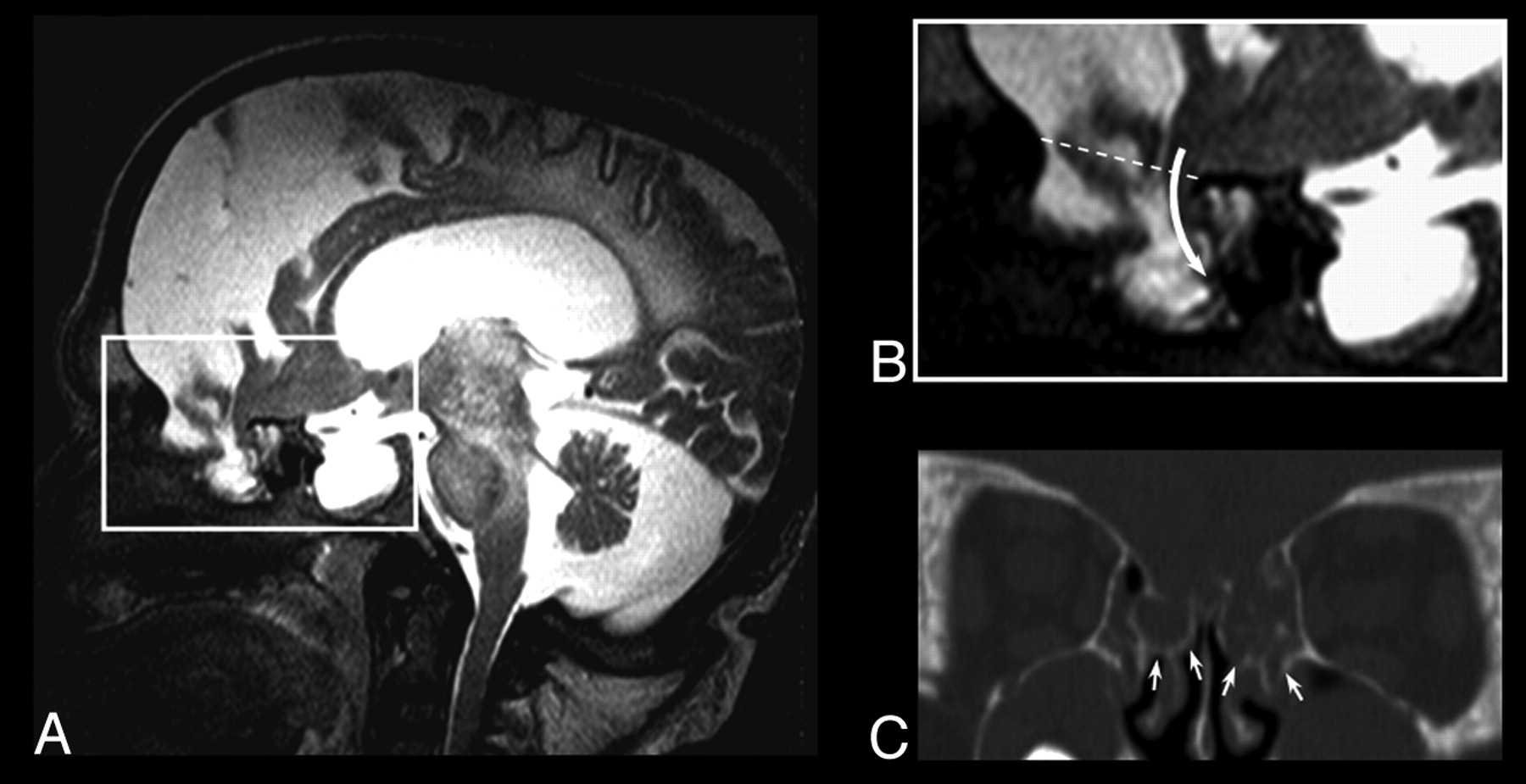
Eight-year-old patient with Hunter syndrome, severe phenotype. A and B, Coronal fluid attenuated inversion recovery images at the level of the anterior portion of the middle cranial fossa disclosing a closed meningoencephalocele on the left side, magnification of A, showing the meningeal pouch with eccentric brain parenchymal herniation (curved arrow); a thin hypointense rim borders the pouch. D, CT coronal image obtained with multiplanar reconstruction technique at the level of C, showing the bone nature (white arrows) of the hypointense rim. E, Axial T2-weighted image revealing a multiloculated large pouch. F, CT volume rendering technique 3D-reconstruction depicting the inner aspect of the bone abnormality (black arrows).
Materials and Methods
Subjects
Of the 36 patients participating in the previous study,12 brain MR imaging studies of 33 subjects (32 males and 1 female, mean age 10.4 years, age range at time of MR imaging examination 2.2–30.8 years) were still available for evaluation.
In all cases, the diagnosis was made by urinary GAGS dosage followed by demonstration of a deficient activity of iduronate-2-sulfatase in cultured fibroblasts or in leukocytes. Multiple sulfatase deficiency was excluded by detection of normal activity of other lysosomial sulfatases. The phenotype was judged as “attenuated” and “severe” by pediatricians with expertise in metabolic disorders according to the classification recently published by Muenzer et al.3
Eleven patients presented with the “attenuated” phenotype (mean age 15 years; age range 4.5–32 years), while 22 were classified as the “severe” phenotype (mean age 10.5 years; age range 2.5–19 years).
Thirty patients were receiving weekly infusions of idursulfase (mean treatment duration 1.4 years, range 0.1–5.5 years) and 4 underwent bone marrow transplantation; 1 patient had both treatments.
According to a previous study on this cohort of patients with Hunter syndrome,12 we recorded the presence of epilepsy and the presence and/or the severity of several MR abnormalities (On-line Table 1).
The scales used for all these parameters are described in Manara et al.12
Image Analysis
The last available brain MR imaging examinations were evaluated by 2 experienced neuroradiologists, blinded to clinical data. Areas of focal brain parenchymal and/or meningeal herniation at the level of chondro-/dermatocranium were recorded and the maximal diameter of the meningeal pouch was measured. When available, CT scans of positive patients were also evaluated.
Statistical Analysis
The association between the presence of parenchymal/meningeal herniation, phenotype, and other neuroradiologic findings were tested through the χ2 or Fisher exact test, and when required, t tests and Mann-Whitney tests. Significance level was set at P < .05.
Results
Nine of 33 patients (27%) presented with a focal herniation of brain parenchyma and/or meninx at the level of the anterior (6 cases) or middle (4 cases) cranial fossa. One patient had skull abnormalities in both the anterior and middle cranial fossa. The middle cranial fossa meningeal pouches were on the left side in 3 of 4 cases; all the anterior cranial fossa pouches were at midline at the level of the lamina cribrosa. The maximal diameter of the pouches ranged between 7 and 36 mm (median 25 mm, mean 24.7 ± 10.5 mm). No meningoencephaloceles were found in the posterior fossa or at the level of the cranial vault.
In all cases, a thin T1- and/or T2-hypointense contour delimiting the meningeal pouch was detected. Brain CT examination was available in 4 positive patients, and in all, this contour resulted in bone tissue in continuity with the skull.
The presence of parenchymal/meningeal herniation showed an association with age (P = .03, t test) and a negative association with the presence of dens hypoplasia (P = .03, Mann-Whitney test), while no correlation was found with the presence or severity of all the other neuroradiologic abnormalities commonly observed in Hunter disease (On-line Table 2). Regarding clinical phenotype or presence of epilepsy, there was no significant difference between patients with Hunter syndrome with and without parenchymal/meningeal herniation. In particular, among the 9 positive Hunter patients, 3 were affected with the severe phenotype and 1 had epilepsy. The small number of bone transplantation and ERT-nontreated patients did not allow a reliable comparative analysis.
Discussion
This study on a large series showed that nearly 1 of 4 Hunter patients presents with a meningeal or brain parenchymal herniation at the level of the anterior or middle cranial fossa.
There was great variability in size and parenchymal involvement, with patients harboring a millimetric middle cranial fossa blistering or large pouches filled with CSF and brain parenchyma, usually prolapsing in an eccentric portion of the cavity (Fig 2). In all patients for whom a CT evaluation was available, a bone wall bordering the meningeal pouch was detected (Figs 1⇓–3). Because the term meningoencephalocele should be restricted to the herniation of brain and/or meningeal tissue through a bone defect, “closed cephalocele” might be a more appropriate definition of these skull abnormalities (Prof. Tortori-Donati, personal communication).

Twenty-year-old patient with Hunter syndrome, attenuated phenotype syndrome. A and C, 12-year-old patient with Hunter syndrome, attenuated phenotype A and B. Sagittal T1-weighted images showing the meningeal pouches at the level of the anterior cranial fossa, mainly filled by CSF (A) or brain parenchyma (B). C, Magnification of A, revealing the abnormal course of the olfactory tract and bulb (arrows) diving toward a depressed lamina cribrosa. D, CT sagittal image obtained with multiplanar reconstruction technique showing the presence of a bone pavement, with foramina for the phila olfactoria (white arrows).

Thirteen-year-old patient with Hunter syndrome, severe phenotype A, Sagittal T2-weighted image showing severe brain atrophy, an enlarged pituitary sella, and a large irregular meningeal pouch at the level of the anterior cranial fossa. B, Magnification of A, showing the brain parenchymal eccentric herniation to the deepest part of the meningeal pouch (curved arrow); the dotted line represents the level of the anterior cranial fossa in normal subjects C, CT coronal image obtained with multiplanar reconstruction technique, revealing the bone defect (white arrows) reaching the middle turbinate on both sides.
The etiopathogenic mechanism that leads to closed cephalocele in Hunter disease remains unknown. In fact, the protrusion of intracranial structures could be due to an increased intracranial pressure and consequent bone remodeling, a primitive defect of skull bone formation, or both.
Increased intracranial hypertension is a relatively common and worrisome complication in MPS, as mucopolysaccharide deposition within Pacconian granulations might lead to communicating hydrocephalus, thus requiring ventricle shunt surgery. In the presence of neurologic deterioration, increased intracranial hypertension and communicating hydrocephalus are diagnosed indirectly by means of neuroradiologic signs, such as the enlargement of subarachnoid spaces, dilation of the third ventricle, and periventricular white matter signal intensity abnormalities. In our study population, we found no correlation between the presence of bone outpouching and the presence or severity of communicating hydrocephalus. This finding might suggest that an increased intracranial pressure does not play a significant role in the origin of closed cephaloceles. Nonetheless, the neuroradiologic and clinical features used for identifying communicating hydrocephalus widely overlap with those indicating brain atrophy, another frequent complication of Hunter syndrome that does not imply intracranial hypertension. The presence of atrophy cases among those with communicating hydrocephalus might therefore have acted as a confounding factor. Moreover, the natural course of intracranial hypertension in this disease has not been defined yet, and transitory intracranial hypertension during early phases of skull formation without neuroradiologic signs of communicating hydrocephalus cannot be ruled out.
Alternatively, a focal primitive defect of skull base might lead to cephaloceles. In fact, the skeletal system is commonly involved in Hunter syndrome, and multiple bone abnormalities are documented by x-ray examination, especially at the level of clavicle, ribs, tibias, skull, and vertebral bodies.2 The storage of GAGS in growing cartilage is thought to cause a bone maturation defect characterized by impaired osteoclast activity and decreased cartilage reabsorption.16 Closed cephaloceles could represent a further manifestation of the pathologic process that affects the whole skeletal system in MPS. Of note, these abnormalities have been found in sites most frequently involved in skull defects in the general population. The complexity of the bone maturation process of these regions likely contributes to their pathogenesis (the middle cranial fossa presents with at least 19 ossification nuclei,14 while the thin lamina cribrosa represents a weak area of the skull due to the several holes for the extraintracranial passage of the phila olfactoria).
The association of closed cephaloceles with age disclosed in this study deserves some caution. We used the last available MR imaging for the evaluation to assure the best imaging quality. The slight prevalence of closed cephaloceles in patients with Hunter syndrome affected by the attenuated phenotype probably outweighs the importance of age, as these patients were significantly older (both for later diagnosis and longer survival) compared with severe-phenotype patients with Hunter syndrome. Also noteworthy, when multiple scans were available, closed cephaloceles were recognizable even in the first MR imaging examination of positive Hunter patients, thus showing no evolution during MR imaging follow-up. The same criterion for MR imaging evaluation likely explains the negative association with dens hypoplasia, which has been shown to be less frequent in the attenuated phenotype and in older patients.12
The clinical significance of closed cephaloceles remains uncertain. Closed cephaloceles did not correlate with the clinical phenotype or with the presence of epilepsy, leaving, at the moment, this peculiar finding among other well-known neuroradiologic abnormalities of MPS—such as the omega-shaped sella, the enlargement of PVS, or the platyspondylia—which are not associated with a poor course of disease. Nonetheless, closed cephaloceles deserve to be described because their presence in the ethmoid bone might interfere with rhinosurgery, and parenchymal brain herniation might lead to a higher risk of epilepsy or cognitive deficits at later ages. Follow-up studies and larger series will help clarify their clinical significance and the possible complications in the long term.
Conclusions
Closed cephalocele is a new, peculiar, and likely overlooked neuroradiologic feature of patients affected by Hunter syndrome. Further studies will define its prevalence in other forms of MPS, the possible evolution over time, as well as the impact/response to early enzyme replacement therapy.
Acknowledgments
The authors thank Chiara Briani, MD, for helpful comments in the preparation of the manuscript.
Appendix
Italian MPS Neuroimaging Study Group
Luca Astarita, MD, Department of Pediatrics, Federico II University, Naples, Italy
Rita Barone, MD, Referral Centre for Inherited Metabolic Diseases, Department of Pediatrics, University of Catania, Catania, Italy
Daniela Concolino, MD, Department of Pediatrics, Magna Graecia University of Catanzaro, Catanzaro, Italy
Alessandro Della Puppa, MD, Neurosurgery Unit, University Hospital of Padua, Padua, Italy
Maja Di Rocco, MD, Unit of Rare Diseases, Department of Pediatrics, G. Gaslini Children's Hospital, Genoa, Italy
Maria Alice Donati, MD, Metabolic and Neuromuscular Unit, Meyer Children Hospital, University of Florence, Florence, Italy
Simona Fecarotta, MD PhD, Department of Pediatrics, Federico II University, Naples, Italy
Agata Fiumara, MD, Referral Centre for Inherited Metabolic Diseases, Department of Pediatrics, University of Catania, Catania, Italy
Francesca Furlan, MD, Rare Metabolic Diseases Unit Fondazione Mariani, Pediatric Department, San Gerardo Hospital, Monza, Italy
Orazio Gabrielli, MD, Division of Pediatrics, Department of Clinical Sciences, Polytechnic University of Marche. Mother and Child Salesi Hospital Ospedali Riuniti, Ancona, Italy
Irene Giovannini, MD, Centre for Rare Diseases, Department of Pediatrics, University of Padua, Padua, Italy
Franco Lilliu, MD, Department of Biomedical Sciences and Biotechnologies, Cagliari, Italy
Rodica Mardari, MD, Neuroradiologic Unit, University Hospital of Padua, Padua, Italy
Elena Procopio, MD, Metabolic and Neuromuscular Unit, Meyer Children Hospital, University of Florence, Florence, Italy
Angelica Rampazzo, MD, Centre for Rare Diseases, Department of Pediatrics, University of Padua, Padua, Italy
Andrea Rossi, MD, Pediatric Neuroradiology Department, G. Gaslini Children's Hospital, Genoa, Italy
Graziolina Sanna, MD, PhD, Department of Biomedical Sciences and Biotechnologies, Cagliari, Italy
Footnotes
Disclosures: Renzo Manara—RELATED: Consulting Fee or Honorarium: Shire, Comments: Honorarium for meeting on Fabry disease 4 years ago; Support for Travel to Meetings for the Study or Other Purposes: Travel to meetings on Hunter and Fabry disease. Rossella Parini—UNRELATED: Board Membership: Genzyme (European Board MPS I Registry), Shire (International Board Hunter Outcome Survey), Comments: 1000 euros; Payment for Lectures (including service on speakers bureaus): Nov 2010—combined treatment with BMT and ERT (Genzyme) session on innovative treatments in the Italian meeting of the Society for Metabolic Diseases and Screening; the meeting was named “Milanopediatria”; April 2011 —brief presentation at the annual Shire Symposium on Lysosomal Diseases, in Madrid, Comments: 1300 euros. Maurizio Scarpa—UNRELATED: Board Membership: Shire HGT, Comments: Chair of the CNS working group, travel and housing; Payment for Lectures (including service on speakers bureaus): Shire HGT, Biomarin, Genzyme, Comments: Honorarium (at meetings); Payment for Development of Educational Presentations: Genzyme, Comments: Honorarium for CME webinar on LSD.
References
- Received May 13, 2011.
- Accepted after revision July 13, 2011.
- © 2012 by American Journal of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.