
Abstract
SUMMARY: Most dementias begin insidiously, developing slowly and generally occurring in the elderly age group. The so-called rapidly progressive dementias constitute a different, diverse collection of conditions, many of which are reversible or treatable. For this reason, prompt identification and assessment of acute and subacute forms of dementia are critical to effective treatment. Numerous other entities within this category of presenile rapid-onset dementias are untreatable such as the prion-related diseases. Neuroimaging aids in the diagnosis and evaluation of many of these rapidly progressive dementias, which include myriad conditions ranging from variations of more common neurodegenerative dementias, such as Alzheimer disease, dementia with Lewy bodies, and frontotemporal dementia; infectious-related dementias such as acquired immune deficiency syndrome dementia; autoimmune and malignancy-related conditions; to toxic and metabolic forms of encephalopathy. This first of a 2-part review will specifically address the ability of MR imaging and ancillary neuroimaging strategies to support the diagnostic evaluation of rapidly progressive dementias due to neurodegenerative causes.
ABBREVIATIONS:
- AD
- Alzheimer disease
- CBD
- corticobasal degeneration
- DLB
- dementia with Lewy bodies
- HD
- Huntington disease
- MSA
- multiple-system atrophy
- PSP
- progressive supranuclear palsy
With the increase of the aging population in the United States, the accurate recognition of dementia types is becoming an important clinical topic. Dementia is marked by cognitive decline, loss of normal memory function, and impairment in judgment without disruption of consciousness. Clinical history-taking, laboratory investigation, and imaging studies should be used with the aim of ascertaining dementia diagnoses related to reversible causes and etiologies amenable to treatment and determining the prognosis for irreversible and progressive illnesses. Unlike their more typical counterparts, rapidly progressive dementias are also more frequently atypical and heterogeneous in their clinical presentation, necessitating the use of additional diagnostic measures, including MR imaging, to improve diagnosis when the clinical situation is tenuous.
Thorough clinical evaluation and laboratory investigation are fundamental for ascertaining progressive dementias, especially those of more acute onset and in presenile patients (those younger than 65 years of age). Laboratory investigations are vital in determining several of the illnesses within the differential diagnosis of a rapid-onset dementia, which, for the purposes of this review, is generally 6 months from onset of symptoms to fulminant dementia. As noted in the differential diagnosis On-line Table, rapidly progressive dementias may present with a wide variety of manifestations and etiologies.
Other forms of rapidly progressive dementia, however, are better characterized by using advanced imaging methods such as MR imaging; 1 study of rapidly progressive dementias noted that MR imaging findings suggested the specific diagnosis in one-third of cases.1 Several excellent clinical reviews of the subject discuss the features of rapidly progressive dementias with supportive laboratory and clinical evaluations, which are abbreviated in this neuroimaging-focused review.2⇓–4
Neurodegenerative Dementias
Most dementias in general are neurodegenerative, most commonly of the Alzheimer type, though there is increasing awareness with greater imaging and more thorough investigations of non-Alzheimer disease–related dementias.2 A review of cases from a referral center for suspected prion disease showed that neurodegenerative disease makes up the largest portion of nonprion diagnoses.4 Conditions within this category generally present with a protracted clinical course of gradual decline in cognitive function. However, genetic variants of several neurodegenerative dementias may present much more rapidly than their prototypical presentations.
Early-Onset Alzheimer Disease.
Unlike its more common form, early-onset AD is characterized by a much shorter time course and greater anatomic and functional alterations.2 Imaging findings in early-onset AD are also slightly different from those in senile AD, with a higher magnitude of atrophy within the occipital and parietal lobes.5 This change is in lieu of the stereotypical hippocampal atrophy; however, the imaging findings common in AD in general may be observed in cases of early onset, which are discussed at greater depth elsewhere.6 Medial temporal lobe atrophy appears to be a great discriminator of AD versus dementia with Lewy bodies and vascular-related cognitive decline.7 Imaging findings within certain subtypes of AD are based on genetic syndromes such as those with a presenilin-1 mutation that manifests white matter changes (hyperintense on FLAIR) atypical of AD.8,9
In AD, MR spectroscopy may aid in the diagnosis, with a notable decrease in N-acetylaspartate and an increase in myo-inositol and choline in patients with AD and those with presymptomatic AD.10,11 While it is outside the scope of this review, positron-emission tomography may offer added diagnostic acumen in recognizing AD with the use of the newly FDA-approved florbetapir F18 injection (Amyvid; Eli Lilly, Washington, DC), which specifically binds to β-amyloid.12 Effort is also being directed at developing appropriate targeted MR imaging contrast agents.
As reviewed at greater length elsewhere, several advanced MR imaging techniques may be useful in making the diagnosis of AD.2,13 DTI is the subject of numerous active research studies and beyond the scope of this focused review. In brief, there is some evidence that DTI shows increased diffusivity within the temporal lobe in AD and may aid in differentiating it from other dementias such as dementia with Lewy bodies.14 Magnetization transfer imaging is one of the latest imaging methods to be applied to AD. Histopathologic information may be inferred from this method in light of the correlation between magnetization transfer ratio and the extent of axonal loss, and these changes may proceed to gross volume loss.2
Dementia with Lewy Bodies.
DLB accounts for a small fraction of presenile-onset dementia syndromes, constituting only approximately 4% of presenile dementias, but its rapidity of decline with an average survival of 3 years places it in the differential diagnosis of rapidly progressive dementias.2,3,15 Frequently, neurologic symptoms in DLB are nonspecific and analogous to other neurodegenerative diseases. Because findings are nonspecific in DLB (such as atrophy within gray matter of the temporal, parietal, and occipital lobes), conventional MR imaging is of greatest benefit in ruling out other causes of rapidly progressive dementia rather than diagnosing DLB specifically.16 Relative preservation of the medial temporal lobe has been one of the few consistent findings of volumetric structural studies; another key difference compared with AD is the preservation of the NAA-to-creatine ratios on MR spectroscopy.17,18 Recent attempts have been made to differentiate DLB from AD, and one such study has noted a lower fractional anisotropy within the white matter of the postcentral gyrus, which correlated with decreased motor function clinically.14 Using DTI, Bozzali et al19 noted decreased fractional anisotropy in many white matter regions, with relatively modest involvement of the temporal lobe. Another recent study by Burton et al16 reported reduced amygdala volume as a possible marker of DLB that correlates with the presence of Lewy bodies on pathology. Although outside the scope of this MR imaging–based review, reduced dopamine transporter levels in DLB, as shown with iodine 123 N-(3-fluoropropyl)-2β-carbomethoxy-3β-(4-iodophenyl)-nortropane SPECT, might assist in the diagnosis of DLB in light of the currently limited role for MR imaging.20
Frontotemporal Dementia.
Frontotemporal dementia, albeit generally with quicker symptom progression than AD, is a rarer form of rapidly progressive dementia.3 Moreover, some clinical attributes differentiate it from many other dementias, with a prominence of personality changes and impaired sociability. Imaging can demonstrate the atrophy for which this condition is named in half of the cases, though early phases of this condition may be missed and imaging is used with the primary purpose of excluding other forms of dementia.2,21 The “knife edge” sign is one imaging finding supportive of frontotemporal dementia on MR imaging and is described as focal atrophy within the anterior temporal lobe at the level of the temporal horn of the lateral ventricle.21,22 One study integrating MR imaging and clinical measures of dementia demonstrated associations between worsening of these rating scales and rates of ventricular expansion and whole-brain volume loss.23
Corticobasal Degeneration.
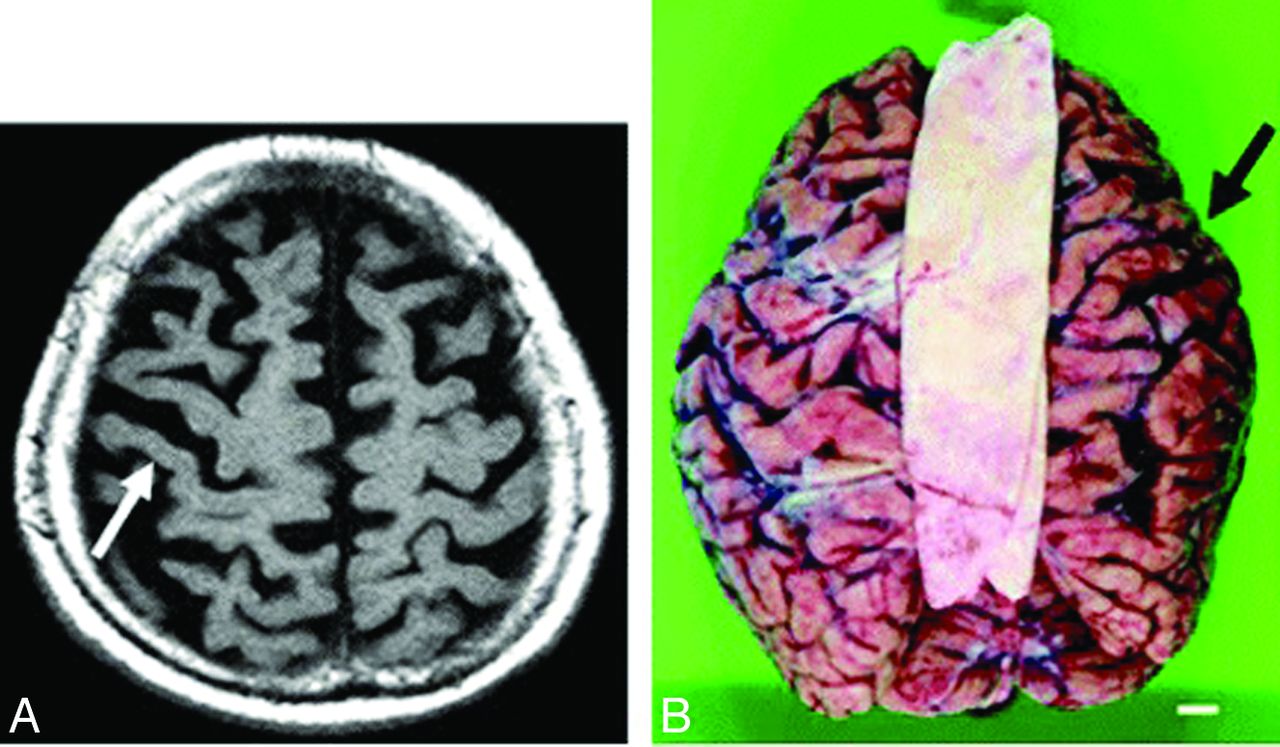
Because CBD may present clinically with progressive dementia and some of the rare neurologic symptoms seen in Creutzfeldt-Jakob disease, such as alien limb and myoclonus, imaging is essential to distinguish these 2 clinically similar entities.3 Patients with CBD may have atrophy of the caudate nucleus, putamen, and some cortical regions (premotor and superior parietal), usually in an asymmetric pattern (Fig 1).24⇓–26 Moreover, cerebral atrophy is substantially greater than that seen in the clinically similar syndrome of progressive supranuclear palsy.25 There may also be increased signal intensity within the subcortical white matter.

Corticobasal degeneration. Imaging performed in an 84-year-old woman with corticobasal degeneration proved on postmortem examination. A, Axial T2-weighted image shows right-side-dominant atrophy, including the central sulcus (arrow). B, Postmortem examination of this patient's brain shows right-frontal-dominant atrophy (arrow).24
Progressive Supranuclear Palsy.
PSP, a tauopathy frequently in the same differential diagnosis as CBD, may manifest early with executive dysfunction or subcortical dementia and then progress to involve the classic clinical presentation of supranuclear gaze palsy and motor symptoms.27 Classically described MR imaging findings typical of PSP were reviewed by Stamelou et al28 and include the classic “humming bird” or “penguin” sign (Fig 2).24 These findings are largely related to atrophy within the midbrain, with involvement of the midbrain, pons, thalamus, superior cerebellar peduncle, and striatum; there may be hypointensity of the putamen on T2-weighted sequences.25,28 Of clinical importance, midbrain atrophy on MR imaging correlates with motor deficits.29 Commensurate with midbrain atrophy, there may be dilation of the third ventricle.28 There may also be T2-weighted hyperintensity of the tegmentum, corresponding to histopathologic evidence of neuronal degeneration in this region; this sign appears to be very specific, but not present in many cases.30

Hummingbird or penguin sign. Imaging performed in a 74-year-old man with PSP. T1-weighted midsagittal image clearly shows atrophy of the midbrain tegmentum (arrow), referred to as the hummingbird or penguin sign. The area of the midbrain tegmentum is 71 mm2.24
Because there is much overlap between PSP and CBD visually on conventional MR imaging, many volumetric studies have attempted to delineate differences between the two, particularly noting the lesser involvement of cortical atrophy in PSP.25,28,31 Also in the literature are less validated visual findings indicative of PSP; a proposed imaging finding is an abnormal superior profile of the midbrain on sagittal T1 images (Fig 3), in which the normal convex profile of the midbrain becomes flattened or concave, which is akin to the subjective hummingbird sign.30 DWI may demonstrate increased putaminal ADC values, a finding that may aid in differentiating PSP from Parkinson disease.32

Abnormal superior profile of the midbrain in progressive supranuclear palsy. Top row: midsagittal T1-weighted spin-echo sections in Parkinson disease (A) and PSP (B and C) show the midbrain region. Bottom row: images with outlined profiles of the upper midbrain, which appears convex in A, linear (flat) in B, and concave in C as a result of midbrain atrophy in PSP.30
Multiple-System Atrophy.
The hallmark feature of this collection of neurodegenerative diseases of unknown cause, multiple-system atrophy, is progressive cerebral atrophy, which has been validated in small groups of patients with serial MR imaging showing rapid atrophy corresponding with the duration of illness.33 Atrophic changes are noted by decreased size on conventional imaging and hypointensity on T2-weighted imaging, particularly within the lower brain stem, middle cerebellar peduncles, cerebellum, and putamen.26,34 In the pons, there is the classic imaging finding of the “hot cross bun sign” (Fig 4).35⇓–37 With this atrophy, there may be a surrounding hyperintense rim seen on T2 images, which may appear before the hot cross bun sign, but it is unfortunately nonspecific, with atrophy being more important in distinguishing MSA from Parkinson disease.36,38 T2* gradient-echo signal loss within the dorsolateral putamen reflective of increased iron deposition is highly specific for MSA, but not always present.39 Serial MR imaging has been used to characterize the atrophy noted within the pons, being particularly good at distinguishing MSA and CBD with a 3-fold greater atrophy rate, both of which can be differentiated on this basis from Parkinson dementia and healthy controls.29

Hot cross bun sign. A 48-year-old woman with spinocerebellar ataxia type 6. T2-weighted fast spin-echo MR image (TR/TE/TI, 3600/102/2 ms) shows bilateral symmetric hyperintensity of atrophic middle cerebellar peduncles. The pons with the “cross sign” and cerebellum are also atrophic, as can be seen in multiple-system atrophy.35
DWI may demonstrate increased diffusivity and ADC within the affected middle cerebellar peduncles, and other authors report decreased fractional anisotropy matching this finding.34,40⇓–42 Putaminal ADC increases likewise exist in MSA, but not Parkinson disease, and these values also correlate with clinical measures.41,43⇓⇓–46 Therefore, DWI and DTI are essential in improving differentiation between MSA and other similar conditions such as PSP and Parkinson disease.
Huntington Disease.
Because of the CAG triplet repeat propagated in an autosomal dominant fashion, clinicians diagnose HD on the basis of genetic testing prompted from a family history of early dementia and extrapyramidal symptoms with the hallmark of chorea. Clinically, there is mounting evidence for preclinical cognitive decline preceding any of the motor symptoms of HD.47 Classic imaging findings reveal atrophy of the caudate nucleus, with concomitant enlargement of the frontal horns of the lateral ventricles.26 This reduced size of the caudate nucleus has been correlated with cognitive performance and precedes motor symptoms by many years (Fig 5).47,48 There may be T2 hyperintensity in the region of the caudate nucleus with this atrophy. Moreover, the basal ganglia may be hypointense on FLAIR and T2-weighted images due to iron deposition, and this finding is thought to constitute an early biomarker for the disease.49 Other advanced MR imaging methods such as MR spectroscopy at a high-field strength may aid in showing changes associated with HD with decreased NAA and creatine within the caudate nucleus and putamen.50
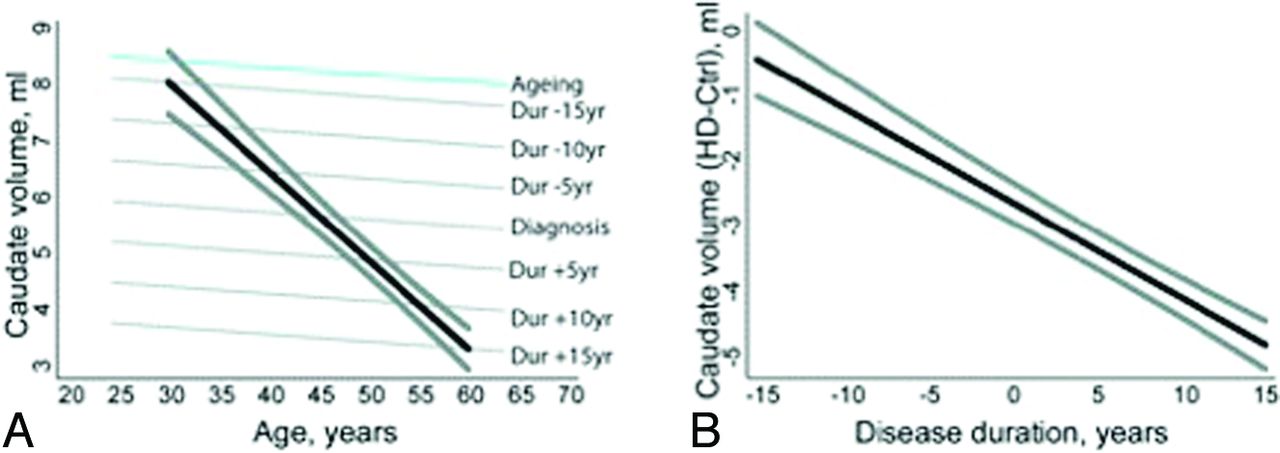
Caudate atrophy in Huntington disease. A, Illustration of the fitted relationship between caudate volume and age in normal aging (blue) and patients with Huntington disease according to disease duration (gray) for male subjects with a total intracranial volume of 1500 mL. The black line shows the predicted trajectory (with the 90% confidence interval [CI] in thick gray) for a patient with HD with motor onset at 45 years of age. B, Fitted difference (with 90% CI) in caudate volume between patients with HD and controls according to disease duration. The difference is statistically significant (P < .05, 1-sided) ≤14 years before motor onset.48
Neuroimaging may prognosticate the onset of disease progression in HD before the development of symptoms. A recent volumetric MR imaging study of patients before the clinical onset of HD symptoms noted that patients who would develop symptoms in 1–4 years following their scans demonstrated significantly smaller striatal volumes when they were still asymptomatic, particularly within the putamen.51 Other authors suggested that MR imaging can detect striatal atrophy even up to 20 years before motor deficits.52 This reduction in striatal volume was previously hypothesized to serve as a biomarker for likely clinical progression of HD.53 Others have defined more global atrophy in cortical structures, perhaps later in the disease process, and these findings are more inconsistent than those involving the striatum.52 From the wealth of clinical studies performed to date, MR imaging has a powerful role in the diagnosis and prognostication of HD.
Conclusions
This first installment of a 2-part review of MR imaging findings in the diverse clinical manifestation of rapidly progressive dementia discussed the use of neuroimaging in diagnosing neurodegenerative conditions, ranging from the common aging-associated dementias to uncommon presentations of less prevalent conditions such as multiple-system atrophy. Newer technologies, such as amyloid imaging, may aid in distinguishing more unusual neurodegenerative causes of rapid cognitive decline and variants of the more common Alzheimer dementia. MR imaging with selective use of DWI, DTI, and MR spectroscopy is essential to narrow the differential diagnosis on the basis of the subtle differences between neurodegenerative dementias, and it plays a key role in establishing the diagnosis. The second part of this review will detail the imaging findings seen in a wide range of other causes of rapidly progressive dementia, including prion, infectious, inflammatory, autoimmune, neoplastic, metabolic, and nutritional conditions.
Indicates open access to non-subscribers at www.ajnr.org
REFERENCES
- © 2014 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}